

Abstract
Objective: Alcohol may have psychomotor stimulant properties during the rising limb of the blood alcohol curve at commonly self-administered doses. Increased heart rate (HR) immediately after alcohol consumption may serve as an indicator or marker of such properties, which appear to be potentially opiate-mediated and dopamine-dependent. Naltrexone, an opiate antagonist, has been used successfully in the treatment of alcoholism and may produce its therapeutic effects through its effects on alcohol metabolism or by blocking alcohol’s rewarding effects. We hypothesized that, if naltrexone blocks the psychomotor stimulant properties of ethanol, then it would decrease or eliminate the HR increase associated with acute alcohol intoxication and that this would be independent of any effect on alcohol metabolism.
Methods: Twenty male subjects were administered placebo and alcohol (1.0 mL 95% USP ethanol/kg body weight) in a laboratory setting on one day and naltrexone (50 mg) and alcohol on another (counterbalanced). We assessed all subjects for a change in HR and for a subjective and behavioural response from 35 to 170 minutes after drug or alcohol administration.
Results: The placebo and alcohol mix produced a significant mean HR increase from baseline (F1,95 = 46.01, p < 0.0001, Cohen’s d = 0.62), while naltrexone and alcohol did not (nonsignificant). The significant effects of naltrexone on blood alcohol level did not account for the effect of naltrexone on alcohol-induced HR change but did account for alterations in subjective and behavioural response to alcohol.
Conclusions: Naltrexone appears to substantially reduce the HR increase that is characteristic of alcohol intoxication. This finding appears to lend moderate support to the notions that, first, naltrexone has differential effects on alcohol reactions and, second, that it specifically blocks the acute psychomotor stimulant properties of alcohol.
Introduction
Psychomotor stimulant or incentive reward effects characterize most substances abused by animals and humans.1 However, ethanol’s tranquilizing, depressant and anxiolytic capacities, particularly at high doses, have obscured the existence of its comparatively transient and individually variable stimulant properties.2,3 Nonetheless, in recent years, the existence of such effects has become evident and their nature more clearly delineated. Alcohol appears to manifest such properties during the relatively short-lived rising limb or peak of the blood alcohol curve, at doses approximating 1 g/kg (1.267 mL/kg).3,4
Psychomotor stimulant effects have generally been attributed to increased dopamine (DA) activity in the mesocorticolimbic system.1 Classic stimulants, such as cocaine and amphetamine, have relatively direct DA effects.1 By contrast, alcohol appears to more directly affect the functioning of the endogenous opioid system and to partially produce its rewarding effects by interacting with this system.5 For this reason, perhaps, administration of opiate agonists and antagonists appear to modulate alcohol consumption. Low doses of morphine have generally been found to prime alcohol consumption at least among animals.6 Conversely, high doses tend to decrease alcohol intake, perhaps by producing satiation.6 Administration of nonselective (naltrexone/naloxone)7,8 and selective opioid receptor antagonists7 tends to decrease voluntary ethanol consumption among animals in most, but not all, experimental situations.6 Similar results have been obtained among humans. A small number of clinical trials have also demonstrated that naltrexone safely9 reduces consumption, craving and likelihood of relapse among detoxified outpatients with alcoholism,10,11 as well as suppressing ad libitum alcohol consumption among heavy drinkers.12 Somewhat attenuated but analogous results have been reported for nalmefene, another opiate antagonist.13
The psychopharmacology of opiate-mediated incentive reward is not entirely understood. However, it appears that the incentive reward properties of opiate agonists are generally dependent on the activation of the DA incentive reward system.2,14 The biological substrate for such opiate-mediated DA activation clearly exists. Tonically active endogenous opiate systems modulate activity in the mesolimbic DA pathway.15 Further, morphine has been shown to stimulate mesolimbic DA release,16 theoretically acting on the mu-receptor located on GABA-interneurons in the ventral tegmental area (VTA).15 In addition, the administration of β-endorphin at incentive rewarding doses, with in vivo micro-dialysis, increases dopamine release in the nucleus accumbens,17 whereas the administration of κ-agonists, which are punishing, decreases such release.18 Alcohol’s opiate-mediated rewarding effects appear similarly DA-dependent,6 and its induction of DA release in the striatum/nucleus accumbens16,19 appears to be mediated by endogenous opiate agonists20 and can be blocked by pretreatment with nonselective and selective opioid receptor antagonists.19
It follows logically from these arguments that individual variation in sober and postethanol endogenous opiate and/or DA system function might underlie, at least partly, individual differences in the tendency to seek out incentive reward from alcohol.6,21 Alcohol-preferring and nonpreferring rodents appear to differ, for genetic reasons, in β-endorphin and enkephalin system activity under basal conditions and after exposure to ethanol.22 Nonalcoholic children with parents who have alcoholism and with extensive family histories of alcoholism appear characterized by similar abnormalities23; they manifested lower basal plasma β-endorphin under basal conditions and large dose-dependent increases when acutely intoxicated. It is possible that dampening of this potential incentive reward response to ethanol may account for the apparently enhanced efficacy of opiate antagonist treatment among people with alcohol dependency who have a history of familial alcoholism.24
Individual variation in putative alcohol-induced opiate or DA incentive reward system activation has proved somewhat difficult to measure — particularly in humans — although some recent work in this regard, depending at least partly on self-report, appears promising.4,25 Measuring HR acceleration after alcohol consumption also appears useful. Under certain limited conditions, an increase in HR from resting baseline apparently reflects activity in the DA incentive reward system.2,14 HR varies, for example, with the intensity of the appetitive motivational qualities of a given stimulus,26 in a dose-dependent manner, when the stimulus is pharmacological.27 Accordingly, HR increase has been used directly in alcohol-challenge studies as a psychophysiological index of response to ethanol-induced incentive reward.14
Our research group has produced a series of studies demonstrating that alcohol-induced HR increase 1) is significantly higher among treatment-seeking severe alcoholics and among nonalcoholic sons of multigenerational men with alcoholism28,29; 2) is positively associated with enhanced mood in the initial stages of ethanol intoxication30; 3) is positively correlated with the production of plasma β-endorphin28; 4) is of longer duration following rapid consumption of alcohol30; (5) is positively correlated with self-report indices and laboratory measures of voluntary alcohol consumption21,31; 6) predicts susceptibility to ethanol’s ability to increase the consolidation of positively valanced events experienced before ethanol ingestion32; 7) is positively associated with reward-seeking, gambling and aggressive personality traits33–35; and (8) is associated with alcohol-induced DA release in the nucleus accumbens.36
One body of research suggests that alcohol’s incentive reward effects are DA-dependent, opiate-mediated and eradicable by opiate antagonists. The other suggests that alcohol’s DA-dependent, opiate-mediated stimulant effects might manifest themselves in increased baseline HR after alcohol administration. We constructed a study at the juncture of the 2 literatures, attempting to determine whether naltrexone could block ethanol’s ability to stimulate baseline HR increase. Our first hypothesis was that alcohol combined with placebo would produce significant increases in resting HR during the rising limb of the blood alcohol curve, but that no such effects would be produced by alcohol in combination with naltrexone. Second, we hypothesized that naltrexone administration combined with alcohol would eliminate alcohol-induced HR increase but would not generally dampen other forms of HR reactivity, such as that attributable to low levels of physical activity. Finally, we hypothesized that the effect of naltrexone on ethanol-induced HR increase would be independent of measures of ethanol metabolism or tolerance.37 To this end, and to investigate the potential effects of naltrexone in modulating alcohol-induced changes in affect, we administered selected measures of mood, subjective intoxication and body sway.
Methods
We recruited 20 young men (mean 22.4, standard deviation [SD] 2.9 yr) who drink socially and who do not use opiate drugs, with scores < 5 on the Michigan Alcoholism Screening Test,38 from advertisements featured in the arts and entertainment and community newspaper freely circulated in the greater Montréal area. We first contacted respondents by telephone. If they indicated their willingness to participate, they were screened in person by a trained interviewer for symptoms of substance abuse and dependence according to the Diagnostic and statistical manual of mental disorders fourth edition (DSM-IV) criteria39 as well as for the presence of other medical conditions for which alcohol or naltrexone administration were contraindicated. All subjects were requested to avoid alcohol for 48 hours and food for 12 hours before each experimental session. The experimental protocol was granted ethical clearance by the Douglas Hospital in Verdun, Quebec, where the study was undertaken.⇓
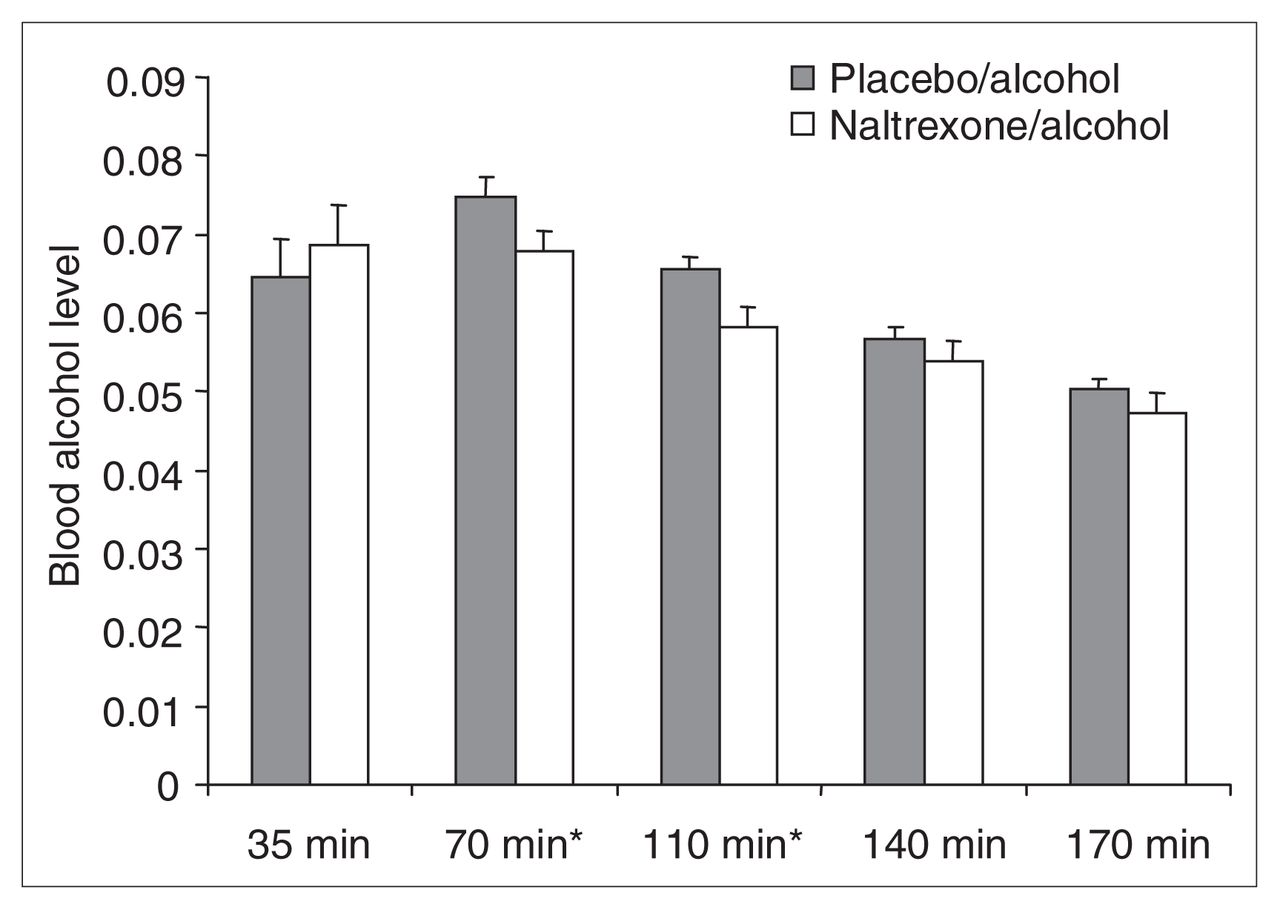
Mean and standard deviation (SD) percent of blood alcohol levels, assessed by breathalyzer, for postalcohol administration onset at 35 min (placebo–alcohol [PA] 0.065; SD 0.021; naltrexone–alcohol [NA] 0.069, SD 0.022), 70 min (PA 0.075, SD 0.011; NA 0.068, SD 0.011), 110 min (PA 0.066, SD 0.007; NA 0.058, SD 0.011), 140 min (PA 0.057, SD 0.007; NA 0.054, SD 0.011) and 170 min (PA 0.050, SD 0.005; NA 0.047, SD 0.011). The difference between drug conditions is significant at p < 0.05 for times marked *.
Each participant received 2 drug combinations in the context of a double-blind, repeated-measures, counterbalanced experimental design. Placebo and alcohol were administered one day, and naltrexone and alcohol were administered on a different day; testing days were separated by at least 72 hours. All subjects arrived at the laboratory at 9:00 am; here they signed an informed consent form, were weighed, were given an initial breathalyzer and underwent urinalysis to ensure the absence of systemic opiate. At 9:30, each subject was asked to relax for 10 minutes. Baseline measures of resting HR and body sway were taken during this period. At 9:40, subjects were administered an opaque capsule containing either naltrexone (50 mg, which has been shown in clinical trials40 to reduce both alcohol consumption and relapse rates in people dependent on alcohol) or cellulose placebo. Seventy-five minutes later, each subject was administered 1.0 mL/kg 95% USP ethanol mixed 1:5 with orange juice. Naltrexone–alcohol dose timing was adjusted in this manner to ensure that blood alcohol level, putative rising limb psychomotor stimulation and naltrexone effects peaked simultaneously.
Alcohol and the ethanol mix were divided into 3 doses, ingested over 15 minutes at the rate of 1 drink per 5-minute period. All subjects were monitored over the course of 3 hours, after the ethanol or drug administration, for alteration in cardiac response. HR was recorded with a Contact Precision Instruments polygraph. Two medi-Trace pellet electrodes placed bilaterally on the subjects’ lower chest were used to detect HR. HR measures were calculated with Psylab computer software for the alcohol drinking interval and then at approximately 35, 70, 110, 140 and 170 minutes after alcohol consumption. We also measured subjective intoxication and body sway. Mean HR measures (for example, 35 minutes after alcohol consumption) represent the average HR for the 10-minute period around a given time point. We estimated the percentage of blood alcohol levels with a breath analysis at the end of each of these measurement periods, except at the alcohol drinking interval (recent presence of alcohol in the mouth made breath analysis at that time impossible). Participants were asked to rest quietly when they were not engaged in activities related to the study. Two subjects, both in the naltrexone–alcohol condition, became ill and were excused from further participation.⇓
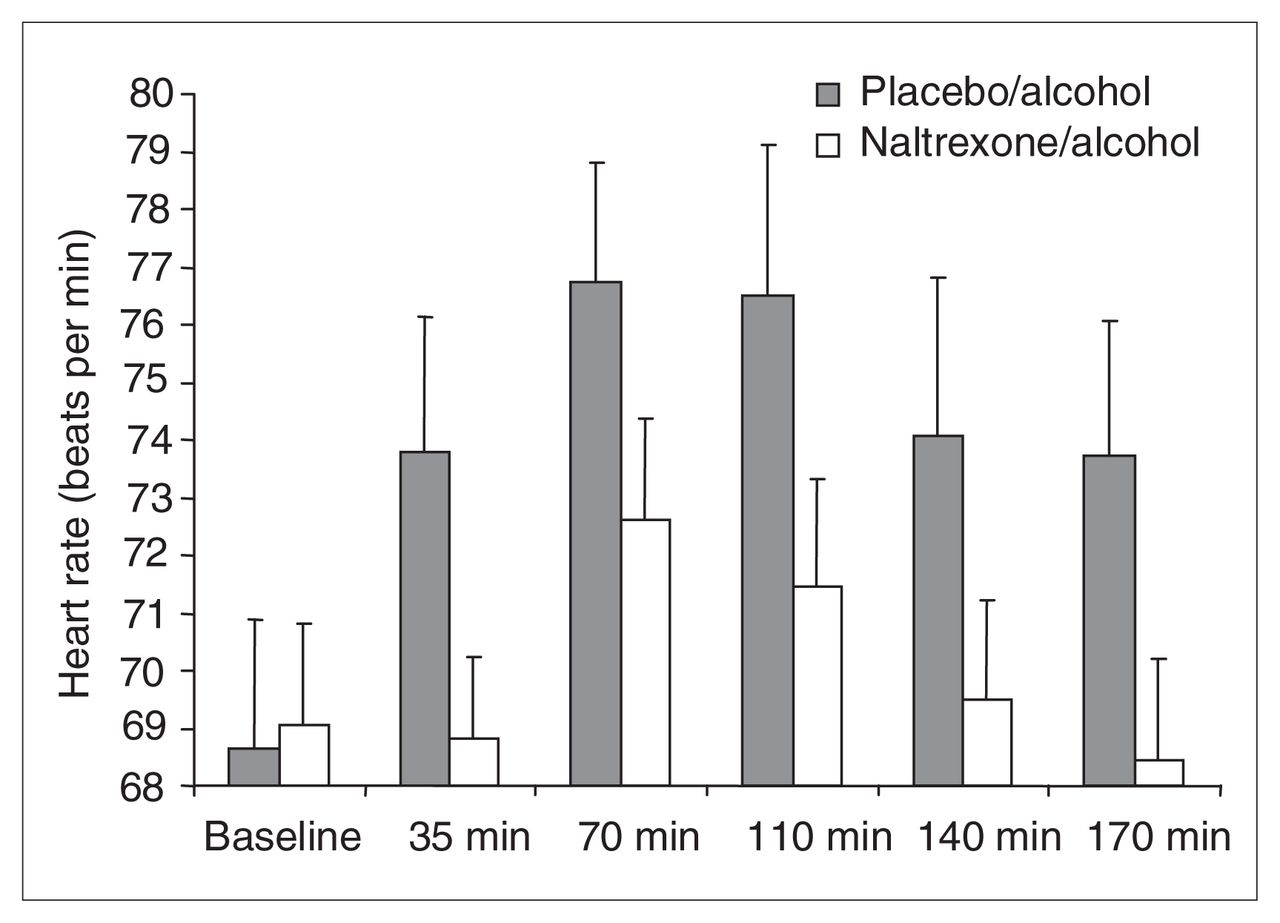
Mean and standard deviation (SD) heart rate (HR) analyses, including baseline measures taken before alcohol or drug administration (placebo–alcohol [PA] 68.65, SD 9.75; naltrexone–alcohol [NA] 69.06, SD 7.66) and measures acquired at 35 min (PA 73.79, SD 10.28; NA 68.82, SD 6.15), 70 min (PA 76.76, SD 8.98; NA 72.61, SD 7.68), 110 min (PA 76.51, SD 11.31; NA 71.47, SD 8.02), 140 min (PA 74.07, SD 12.02; NA 69.48, SD 7.58) and 170 min (PA 73.72, SD 10.23; NA 68.44, SD 7.67) after alcohol administration onset. The figure reveals a significant drug condition × time interaction, decomposed as follows: PA HR, averaged across 35–170 min, is significantly greater than preplacebo–prealcohol baseline HR. NA HR is not. PA HR, averaged across 35–170 min, is significantly greater than averaged NA HR.
Alcohol-induced changes in mood were assessed with the Profile of Mood States (POMS)–Bipolar Form.41 This well-validated paper-and-pencil scale, designed as an inventory of mood states in healthy and in mentally ill populations, appears sensitive to changes in mood states across several dimensions.41 Six bipolar dimensions of mood were assessed with the following subscales, as outlined by Lorr41: composed–anxious (C-A), elated–depressed (E-D), energetic–tired (E-T), agreeable–hostile (A-H), clearheaded–confused (C-C) and confident–unsure (C-U). Each dimension of mood was measured after a sober resting baseline period and then after each alcohol-intoxicated resting HR recording period, at approximately 35, 70, 110 and 140 minutes after alcohol consumption.
Subjective intoxication ratings were assessed with the Subjective High Assessment Scale (SHAS).42 The SHAS comprises 17 items that detail the subjective effects of a given drug (e.g., dizzy, high, sleepy, clumsy). Subjects are asked to rate on a 36-point Likert scale the degree to which they feel minimal (normal = 1) or maximal (extremely = 37) drug effects. An item analysis of the SHAS indicates that the scale reflects both positive (feel high) and negative (feel drowsy) alcohol effects but appears strongly weighted toward negative effects. People prone to heavier drinking and at apparent risk for the development of alcohol problems typically appear less sensitive to the subjective effects of alcohol, according to this scale,43 particularly with regard to negative effects. A recent multivariate analysis indicated that a factor comprised mostly of negative items measured at 60–100 minutes after alcohol consumption (descending limb) most optimally identified individuals at risk for the development of alcohol dependence.43 We assessed subjective responses to alcohol 4 times along the blood alcohol curve, at approximately 35, 70, 110 and 140 minutes after alcohol consumption.
We measured body sway according to the procedure described by Schuckit37 and originally developed by Moskowitz, Daily and Henderson.44 Two velcro harnesses were placed around the torso of each participant; the subjects were then asked to stand as still as possible for 1 minute with their eyes open, feet together and hands at their sides. Two ropes extending perpendicularly from the front of the participant’s chest and the left side of his torso passed over a separate pulley and were attached to weights hanging from the pulley. An electronic counter measured the number of 0.5-cm intervals of movement that the ropes produced on the pulley rotors. For each body sway recording, 3 separate 1-minute recordings were taken, separated by 30 seconds of rest. Body sway scores for each time period were calculated as the average amount of movement across both movement directions (anterior/posterior and lateral), across the 3, one-minute trials. This behavioural measure of response to alcohol has been shown to distinguish between individuals at different risk for alcoholism: genetically predisposed individuals sway less following the ingestion of moderate or high doses of alcohol, compared with those with no such predisposition.37
At the end of the experimental protocol, subjects were debriefed and paid $5.00 an hour for their participation.
Results
We compared preplacebo–alcohol and prenaltrexone–alcohol administration, using 1-way repeated-measures analysis of variance (ANOVA), where such measures were available, to ensure that baselines across the 2 sessions were equivalent. We then employed 2-way repeated measures ANOVA, using drug condition (placebo–alcohol/naltrexone–alcohol) and time period as the repeated measures for blood alcohol level, HR, mood, SHAS and body sway. Because significant blood alcohol level differences between the 2 drug conditions emerged, we duplicated all of the significant analyses for HR, mood, SHAS and body sway, using residual measures derived from regression and controlling for blood alcohol level, to determine which effects were attributable to simple blood alcohol level differences. Where significant effects emerged, we conducted a more detailed analysis in accordance with the hypothesis pertaining to each measure.
All baseline measures for blood alcohol level measured zero. We conducted a 2-way ANOVA (drug condition × time) on blood alcohol levels, with drug condition (placebo–alcohol or naltrexone–alcohol) and time (35, 70, 110, 140 and 170 min) as repeated measures.
This analysis revealed no significant main effect for drug condition (Huynh-Feldt Epsilon-corrected F1,19 = 2.06, p < 0.17), a significant effect for time (Huynh-Feldt Epsilon-corrected F1.37,26.10 = 17.42, p < 0.0001) and a significant interaction (Huynh-Feldt Epsilon-corrected F1.83,34.69 = 3.99, p < 0.031).
Tests of simple main effects for drug condition revealed nonsignificant effects on blood alcohol level at 35 (F1,19 = 1.20, p < 0.287), 140 (F1,19 = 1.02, p < 0.325) and 170 (F1,19 = 1.28, p < 0.272) minutes but significant effects at 70 (F1,19 = 4.85, p < 0.040) and 110 (F1,19 = 8.25, p < 0.010) minutes, indicating that blood alcohol levels in the naltrexone–alcohol condition were lower during the latter time periods.
Heart rate: drug condition and time effects
No differences emerged for HR when we compared baseline measures before alcohol–naltrexone and alcohol–placebo administration with 1-way repeated-measures ANOVA. We conducted a 2-way ANOVA (drug condition × time) on HR with drug condition (placebo–alcohol or naltrexone–alcohol) and time periods (baseline, 35, 70, 110, 140 and 170 min) as repeated measures. This analysis revealed a nearly significant effect of drug (Huynh-Feldt Epsilon-corrected F1,19 = 4.22, p < 0.054), a significant effect of time (Huynh-Feldt Epsilon-corrected F3.95,75.00 = 9.04, p < 0.0001) and a significant drug-by-time interaction (Huynh-Feldt Epsilon-corrected F5,95 = 3.25, p < 0.009).
We investigated the possibility that these effects were due to blood alcohol differences between drug conditions by repeating the analyses, using HR measures at 35–170 minutes and correcting for blood alcohol level. This correction was undertaken by regressing the HR onto the blood alcohol level at each time point to obtain a residual and by using the data points obtained in place of a raw HR. This procedure was undertaken separately for the placebo–alcohol and naltrexone–alcohol groups. With Pearson’s correlation coefficient, the average correlation for residualized and raw data points was r = 0.97 (SD 0.04), with a minimum r = 0.88 and a maximum r = 0.99.
These analyses revealed a significant effect of drug (Huynh-Feldt Epsilon-corrected F1,19 = 5.00, p < 0.037), a significant effect of time (Huynh-Feldt Epsilon-corrected F4.64,88.19 = 8.16, p < 0.0001) and a significant drug-by-time interaction (Huynh-Feldt Epsilon-corrected F5,95 = 2.90, p < 0.018). Drug effects on alcohol-induced changes in HR were therefore revealed as essentially independent of the effects of naltrexone on blood alcohol levels.
Planned contrasts, conducted on the blood alcohol level uncorrected data, revealed that HR for the placebo–alcohol condition (mean 74.97, SD 9.99), averaged across 35–170 minutes was significantly higher than preplacebo–alcohol baseline HR (mean 68.65, SD 9.75) (F1,95 = 46.01, p < 0.0001, Cohen’s d = 0.62), whereas the naltrexone–alcohol condition HR (mean 70.16, SD 6.78), averaged across 35–170 min, was not greater than the prenaltrexone–alcohol baseline HR (mean 69.06, SD 7.66) (F1,95 = 1.40, ns, Cohen’s d = 0.18).
Additional planned contrasts revealed that HR for the placebo–alcohol condition averaged across the 5 postalcohol consumption time periods (mean 74.97, SD 9.99) was significantly higher than HR for the naltrexone–alcohol condition, similarly averaged (mean = 70.16, SD 6.78) (F1,95 = 79.81, p < 0.0001; Cohen’s d = 0.58). Contrasts conducted with HR measures corrected for blood alcohol level produced nearly identical results.
Heart rate: alcohol drinking interval effects
To explore the effects of naltrexone on HR increase that are caused by factors other than alcohol consumption (in this case, the physical movements associated with beverage consumption itself), we obtained subjects’ HR during the alcohol drinking interval in both drug conditions and compared them with the baseline measures (as portrayed in Fig. 3).

Mean and standard deviation (SD) of heart rate (HR) for placebo–alcohol (PA) and naltrexone–alcohol (NA) conditions at predrug/prealcohol baseline and during the alcohol drinking interval. HR analyses included baseline measures taken before alcohol or drug administration (PA 68.65, SD 9.75; NA 69.06, SD 7.66) and measures acquired during the alcohol drinking interval (PA 75.30, SD 10.36; NA 75.81, SD 7.13). The figure reveals a significant main effect of time but no effect of drug condition and no interaction. The physical activity associated with the act of drinking itself therefore appears to have raised HR equally in both groups.
We conducted a 2-way ANOVA (drug condition × time) on HR with drug condition (placebo–alcohol or naltrexone–alcohol) and time periods (baseline and alcohol drinking interval) as repeated measures. This analysis revealed no effect of drug condition (Huynh-Feldt Epsilon-corrected F1,19 = 0.08, p < 0.78), a significant main effect of time period (Huynh-Feldt Epsilon-corrected F1,19 = 85.02, p < 0.0001) and no interaction (Huynh-Feldt Epsilon-corrected F1,19 = 0.00, p < 0.950). Thus HR in both groups increased equally in response to the minor physical demand of consuming alcohol.
No differences emerged for any of the mood scales when baseline measures before administration of alcohol–naltrexone and alcohol–placebo were compared with 1-way repeated-measures ANOVA. Two-way ANOVAs with repeated measures (2 drug conditions × 5 time periods: baseline and 35, 70, 110 and 140 min) conducted separately for each POMS mood subscale also revealed no significant main effects or interactions.
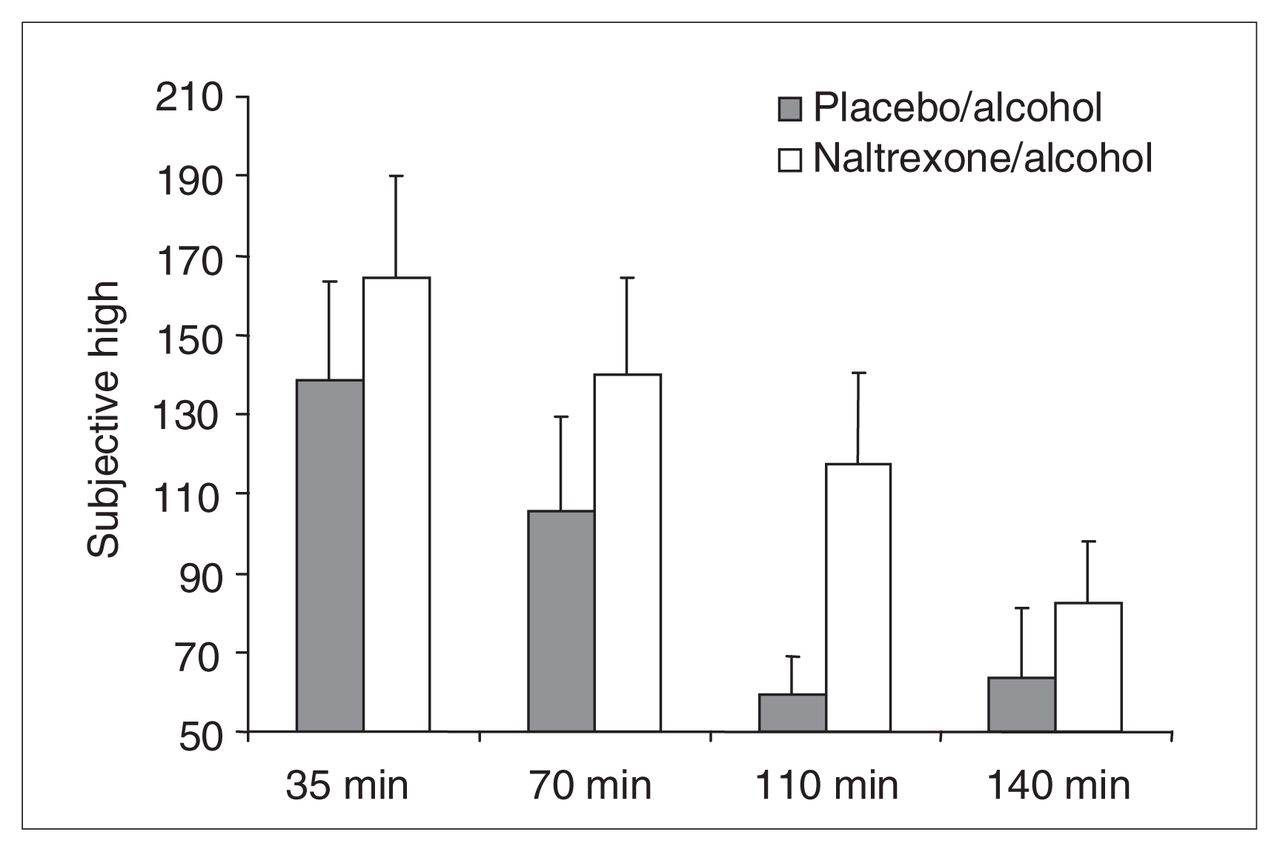
No baseline measures were taken for subjective intoxication. A 2-way ANOVA (drug condition × time) was conducted on SHAS total scores with drug condition (placebo–alcohol or naltrexone–alcohol) and time period as repeated measures. This analysis revealed a significant main effect of drug condition (Huynh-Feldt Epsilon-corrected F1,18 = 11.74, p < 0.003) and a significant effect of time period (Huynh-Feldt Epsilon-corrected F2.54, 45.8 = 18.67, p < 0.0001), but no interaction (Huynh-Feldt Epsilon-corrected F2.90, 52.27 = 1.06, p < 0.37). These results, portrayed in Figure 4, indicate that subjective intoxication ratings were comparatively exacerbated in the naltrexone–alcohol condition.
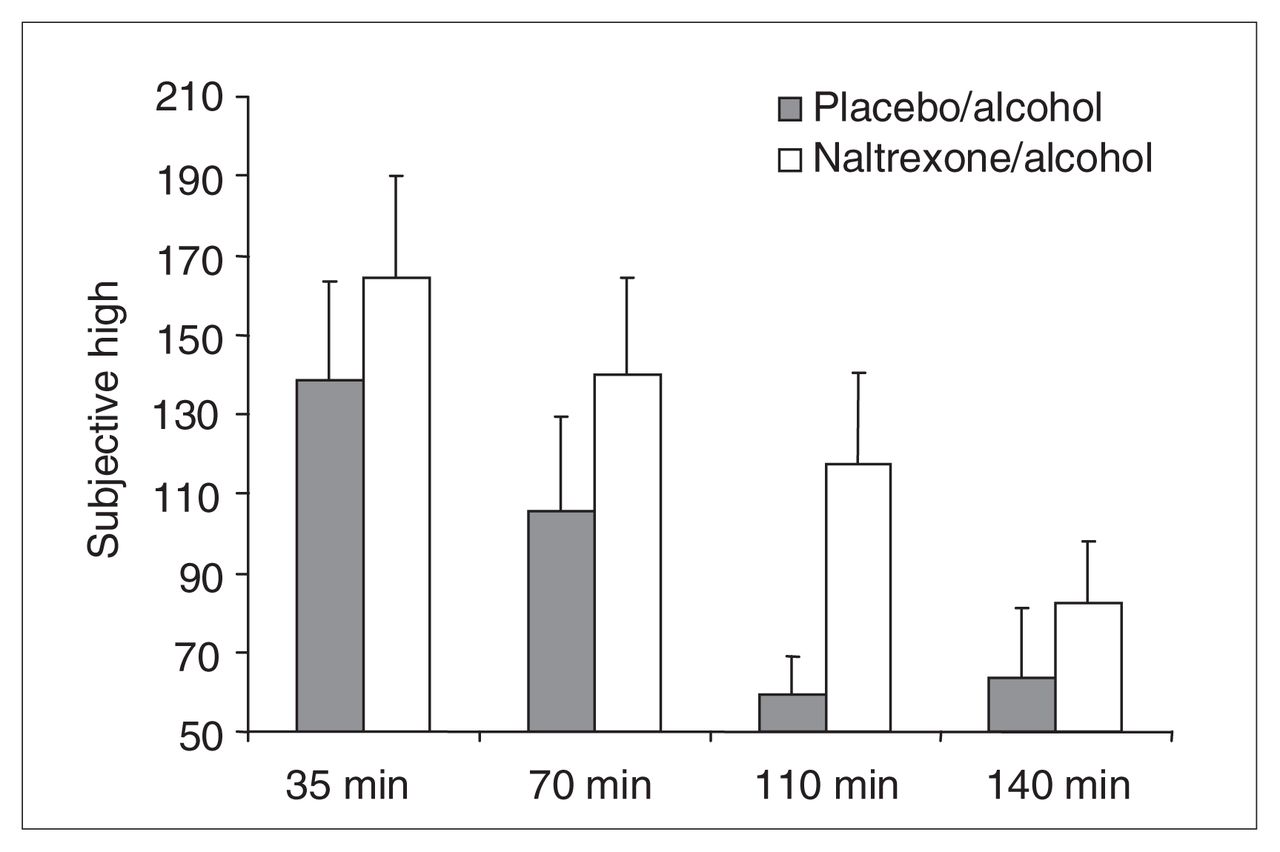
Subjective High Assessment Scale (SHAS) scores for the following time postalcohol administration onset: 35 min (placebo–alcohol [PA] 138.6, SD 109.0; naltrexone–alcohol [NA] 164.3, SD 112.8), 70 min (PA 105.6, SD 104.8; NA 139.9, SD 106.7), 110 min (PA 59.5, SD 41.4; NA 117.5, SD 100.1) and 140 min (PA 63.7, SD 76.0; NA 82.6, SD 65.9). The figure reveals a significant main effect of drug (SHAS score elevated in the NA condition) and time (SHAS score decreased significantly from 35 to 140 minutes) but no interaction. These effects disappeared when the effect of naltrexone on blood alcohol levels was statistically eliminated.
The possibility that these effects were owing to the differences in blood alcohol levels between drug conditions was investigated by repeating the analyses with SHAS measures at 35–140 minutes and correcting by regression for blood alcohol level (as in the HR analysis). Correlations for residualized and raw data points averaged r = 0.978, with a minimum r = 0.91 and a maximum r = 1.00. When the repeated measures ANOVA was repeated on these residual scores, nonsignificant drug and time effects emerged (p > 0.90), as well as no interaction (p > 0.90). Drug effects on alcohol-induced changes in subjective intoxication ratings therefore appeared entirely mediated by the effects of naltrexone on blood alcohol levels.
Body sway
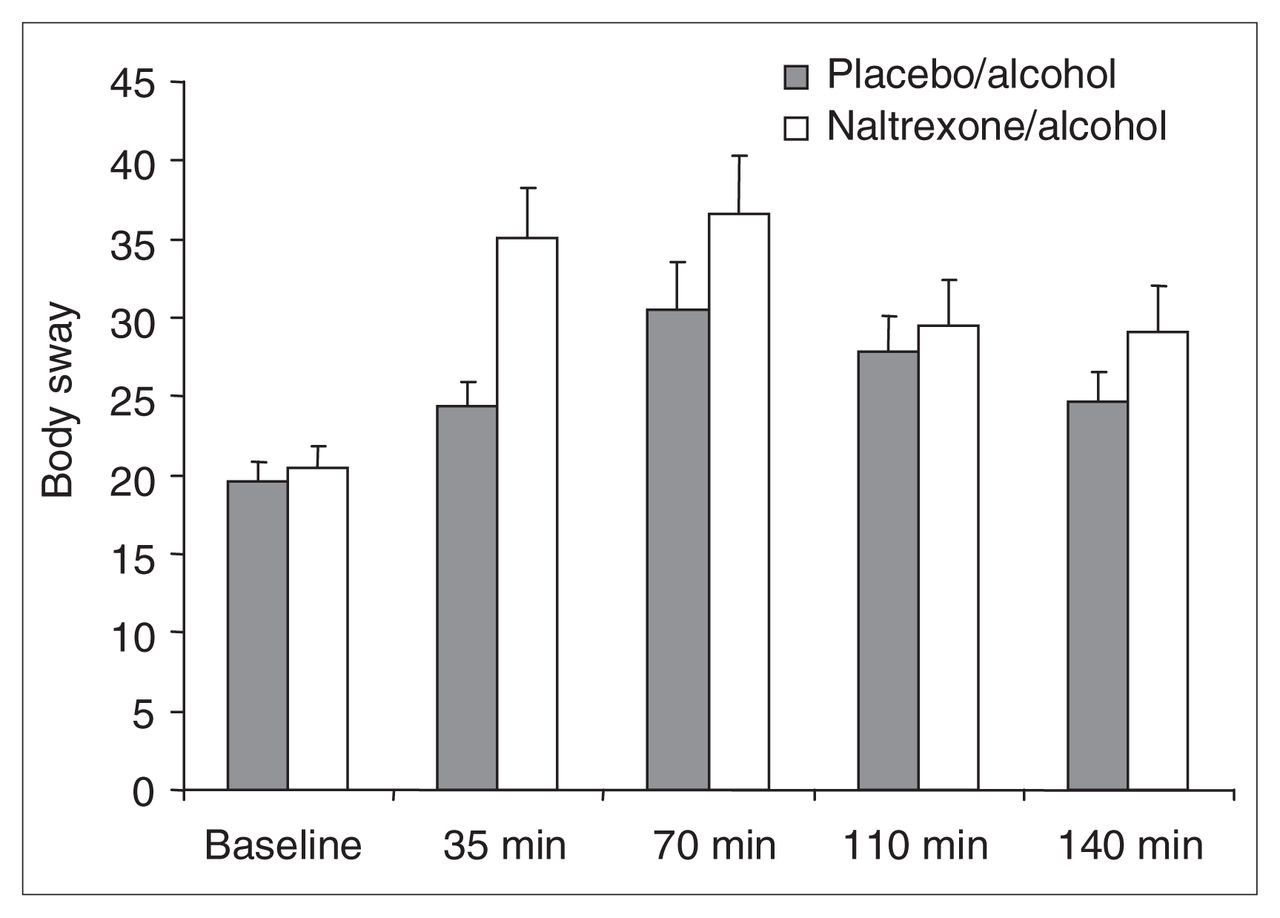
No differences emerged when we compared baseline measures of body sway before alcohol–naltrexone and alcohol–placebo administration, with 1-way repeated measures ANOVA. Body sway measures for the 2 drug conditions were compared across baseline and at 35, 70, 110 and 140 minutes (Fig. 5). Two-way ANOVA, with drug condition and time period as repeated measures, revealed a near-significant effect of drug (Huynh-Feldt Epsilon-corrected F1,18 = 4.00, p < 0.061), a significant effect of time (Huynh-Feldt Epsilon-corrected F3.74,67.35 = 14.52, p < 0.0001) and a trend toward a significant drug-by-time interaction (Huynh-Feldt Epsilon-corrected F2.90,63.16 = 2.07, p < 0.10). These results indicate that alcohol-induced body sway was somewhat exacerbated in the naltrexone–alcohol condition, compared with the placebo–alcohol condition (Fig. 5).

Mean and standard deviation (SD) of body sway scores at baseline and at 35, 70, 110 and 140 minutes postalcohol consumption onset: baseline (placebo–alcohol [PA] 19.6, SD 5.3; naltrexone–alcohol [NA] 20.5, SD 5.8), 35 min (PA 24.4, SD 6.7; NA 35.1, SD 13.22), 70 min (PA 30.5, SD 13.0; NA 36.6, SD 15.6), 110 min (PA 27.9, SD 9.9; NA 29.4, SD 12.5), and 140 min (PA 24.7, SD 8.2; NA 29.1, SD 12.1). The figure reveals a significant main effect of drug, a significant effect of time and a significant interaction. These effects were no longer significant when the effect of naltrexone on blood alcohol level was statistically eliminated.
We investigated the possibility that these effects were caused by the blood alcohol level differences between drug conditions by repeating the analyses, using body sway measures at 35–140 minutes and correcting by regression for blood alcohol level (as in the previous HR and SHAS analyses). Using Pearson’s correlation coefficient, correlations for residualized and raw data points averaged r = 0.978, with a minimum r = 0.93 and a maximum r = 1.00. When the repeated-measures ANOVA was repeated on these residual scores, nonsignificant drug and time effects emerged (p > 0.90), as well as no interaction (p > 0.95). Drug effects on alcohol-induced changes in body sway therefore appeared entirely mediated by the effects of naltrexone on blood alcohol levels.
Discussion
The present study demonstrated that a placebo–alcohol combination produces higher peak blood alcohol levels than does a naltrexone–alcohol combination — a phenomenon that appears potentially related to naltrexone’s capacity to reduce alcohol absorption (demonstrated in rats by Linseman and Le45); this, in turn, appears to be related to opiate modulation of gastric emptying rate and intestinal motility. Nonetheless, Linseman and Le45 concluded that the documented effect of naltrexone on alcohol consumption was probably not attributable to its effects on alcohol absorption.
More importantly, the study demonstrated that a placebo–alcohol combination is capable of producing a robust HR increase, compared with baseline levels before drug administration, whereas a naltrexone–alcohol combination is not. Further, when a control variable was instituted for the blood alcohol differences, there was no change. This result should be interpreted in light of the third demonstration of this study — that naltrexone does not appear to interfere with cardiac reactivity, per se (as elicited during the act of consuming alcohol) — and in light of the fact that naltrexone itself has no effect on baseline resting HR at the dose administered in this study (at least among abstinent alcoholics).46 This is not to say that the effect of naltrexone on blood alcohol level is without consequence. Significant placebo–alcohol versus naltrexone–alcohol differences did emerge for SHAS and body sway, but these differences disappeared when group variation in blood alcohol level was controlled statistically.
Why did the HR increase that is characteristic of placebo–alcohol mix fail to appear in the naltrexone–alcohol condition? Naltrexone modulation of opiate-mediated dopaminergic psychomotor stimulation (indexed by HR increase) appears to be a likely cause. The fact that HR stayed high in the placebo–alcohol condition long after blood alcohol peaked — and, therefore, long into the descending limb of the blood alcohol curve — might seem to mitigate against such a hypothesis. However, it appears that the rate of blood alcohol level increase during the ascending limb is associated with the same processes that produce subjective feelings of positive affect (or some combination of rate and absolute level attained), rather than simply an absolute blood alcohol level attained or an absolute duration of elevated blood alcohol level.30
The findings that the subjective, behavioural and psychophysiological effects of alcohol could be differentiated and the hypothesis that they appear mediated by opiate mechanisms different from those modulating the alcohol-induced HR increase appears consistent with research demonstrating a dissociation between subjective (i.e., liking) and reinforcing (i.e., wanting) properties of rewarding stimuli,47 or the distinction between subjective sensitivity to drug effects and motivation for drug self-administration.48,49 We were not able to test whether alcohol-induced HR increase was associated with sensitivity to alcohol-induced incentive reward or actual drinking behaviour, per se, in the present study. However, much of the research conducted by our group as well as earlier research lend direct credence to this idea.
It is important to mention the results of McCaul and colleagues50 in the context of this study. These authors also explored the effects of naltrexone on cardiac and subjective reponses to alcohol. Although they reported a trend toward an interaction between a high dose of naltrexone and a high dose of alcohol on subjective ratings of liking for alcohol, their results more strongly supported an overall effect of naltrexone on subjective ratings across all doses of alcohol consumed, including placebo dose; that is, naltrexone was associated with lower subjective liking measures overall, regardless of alcohol dose. Curiously, McCaul and colleagues’ study did not yield an effect of naltrexone on blood alcohol level, which is inconsistent with the results of the current study and of previous studies with rodents.46 These differences may be due to the fact that we tested the effects of an acute dose of naltrexone combined with alcohol 75 minutes after naltrexone was ingested, whereas McCaul and colleagues administered similar doses of naltrexone the night before alcohol consumption. It is possible that the effects of naltrexone on alcohol absorption are more acute and short-lasting than are the effects of naltrexone on centrally mediated opiate mechanisms. Indeed, opiate mechanisms have been implicated in peripherally occurring alterations in alcohol absorption46 and in the centrally mediated development of sensitization of the locomotor response to alcohol.51
We should note 2 fundamental limitations to this study. First, because of limited resources and because alcohol abuse is much more common among men,52 we targeted males as our population of interest. However, men and women may differ in their short-40 and long-term53 response to alcohol and to other putatively stimulant drugs. Consequently, the generalizability of our results to women has not yet been established. Second, we only examined the acute effects of one dose of naltrexone on alcohol intoxication, and we held the time after naltrexone ingestion to alcohol ingestion constant. People taking naltrexone for alcohol dependence ingest it on a daily basis and drink variable doses of alcohol at variable times afterward. It would be interesting to determine how long after naltrexone administration the alcohol-induced HR increase dampening effect lasts and whether this varies with naltrexone and/or alcohol dose. It would also be interesting to determine whether individuals who are characterized by sensitivity to naltrexone’s dampening effect on alcohol-induced HR increase are also those who benefit from its long-term clinical effect (assuming that such sensitivity varies substantively across individuals being treated for alcoholism).
Despite these limitations, we propose that naltrexone’s effectiveness as a treatment for alcoholism might be directly related to its psychomotor stimulant blocking effects — at least among men. The dependence of such stimulant effects on the presence of mu-opioid receptors in rats has recently been demonstrated.54 When used to treat alcoholism, naltrexone appears to induce classical extinction, as might be expected if opioid-mediating stimulant blocking were its mechanism of action.55,56 Indeed, opiate antagonists typically have little effect when first administered,55,56 progressively decrease alcohol consumption over time (at least among rats56,57) and appear to block ethanol’s incentive reward or “euphoric” effect.58 Further, opiate antagonist treatment decreases alcohol consumption most markedly for rats55 that drank while under opiate-antagonist influence (although this effect has not been replicated in people with alcoholism59). Finally, drinking reduction that is attendant on naltrexone treatment persists after discontinuation of active medication.56,60 This may indicate, at least hypothetically, that some more-or-less permanent learning has taken place, with regard to the now less desirable effects of alcohol and that naltrexone effects on alcohol-induced psychomotor stimulation, possibly in combination with its mildly aversive effect (mediated by its influence on alcohol absorption), might be responsible for such learning.
Acknowledgements
This research was funded by the Alcoholic Beverages Medical Research Foundation and the Medical Research Council of Canada.
Footnotes
Medical subject headings: alcohol; heart rate; naltrexone; stimulant.
Competing interests: None declared.
Contributors: All authors contributed to the conception and design of the article, its analysis and interpretation. All authors wrote the article, and all authors gave approval for the final version of the article to be published.
- Received June 3, 2004.
- Revision received July 11, 2005.
- Revision received June 21, 2006.
- Accepted June 21, 2006.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.