

Abstract
Smoking is the leading cause of preventable illness in the world today. Prenatal cigarette smoke exposure (PCSE) is a particularly insidious form because so many of its associated health effects befall the unborn child and produce behavioural outcomes that manifest themselves only years later. Among these are the associations between PCSE and conduct disorders, which have been mostly ascribed to the deleterious effects of nicotine on the fetal brain. Here we hypothesize that inhibition of brain monoamine oxidase (MAO) during fetal brain development, secondary to maternal cigarette smoking and in addition to nicotine, is a likely contributor to this association. MAOs play a central role in monoaminergic balance in the brain, and their inhibition during fetal development — but not during adult life — is known to result in an aggressive phenotype in laboratory animals. This paper provides theoretical and experimental support for the notion that cigarette smoke–induced inhibition of MAO in the fetal brain, particularly when it occurs in combination with polymorphisms in the MAOA gene that lead to lower enzyme concentration in the brain, may result in brain morphologic and functional changes that enhance the risk of irritability, poor self-control and aggression in the offspring. It also encourages research to evaluate whether the interaction of smoking exposure during fetal development and MAOA genotype increases the risk for conduct disorder over that incurred by mere fetal exposure to tobacco smoke.
Introduction
One in 5 Americans — roughly 45 million adults — smoke,1 and close to 40% are women. Estimates indicate that 1 in 10 pregnant mothers smoke cigarettes, which is a rate far higher than the 1% goal proposed by Healthy People 2010.2
Smoking during pregnancy has been identified as the single most preventable cause of illness and death among mothers and infants.3 This behaviour has been linked to many deleterious effects that put the newborn at increased risk for adverse health outcomes. For example, maternal smoking during pregnancy is dose-dependently associated with low birth weight and reduced growth of the fetal head,4 the harbingers of impaired cognition that could help explain the observed associations between prenatal cigarette smoke exposure (PCSE) and increased risks for attention-deficit hyperactivity disorder, antisocial behaviours and neurocognitive deficits in the offspring (see Neuman and colleagues5 for a list of references). However, the mechanisms underlying such long-term effects in the offspring have been difficult to establish, likely because of the confounding effects of many genetic, epigenetic, developmental and environmental factors, whose combined actions help train these associations. Our goal is not to present a comprehensive review of these factors and associations but to focus on the type of disruptive behaviours categorized as conduct disorders.
Conduct disorders encompass a group of aggressive and nonaggressive behavioural and emotional syndromes in children and adolescents who have difficulty following rules and behaving in socially acceptable ways. These disorders affect between 3% and 8% of preadolescent and adolescent boys and are more common in boys than in girls.6 According to some estimates, smoking during early pregnancy may account for up to 25% of externalizing (aggressive) behaviours, which include conduct disorders.6 In the past 15 years, numerous epidemiologic surveys have documented robust links between PCSE and externalizing conduct disorders.7–17 An association with aggressive behaviours was also suggested by a large longitudinal Danish study that found that maternal smoking predicted persistent criminal outcome in male offspring even after correcting for parental characteristics or perinatal complications.18 Collectively, the specificity of these effects and their timing strongly suggest that maternal smoking may contribute to the development of conduct disorders including aggressive behaviours, a hypothesis that is supported by preclinical studies. 19 Disturbingly, the association could be self-perpetuating because PCSE appears to be a risk factor for nicotine dependence later in life,20 and a childhood history of conduct disorder may be a risk factor for maternal smoking during pregnancy. 21 Indeed, it is possible that a significant overlap exists between the genes that confer vulnerability to conduct disorders and those that modulate smoking behaviours.
The challenging issue is to determine whether exposure to tobacco smoke while in utero can cause antisocial behaviours or whether it merely indicates the presence of other risk factors (e.g., common genetic vulnerabilities, postnatal exposure to cigarette smoke) that modulate or lead to this class of psychiatric disorders in the offspring. Included among these factors are a mother’s subsequent smoking habits,22 a mother’s history of antisocial behaviour17,23 and depression symptoms17 and the family’s socioeconomic status10,17 or loading for alcohol dependence.24 Moreover, in one study correcting for parental depression and antisocial behaviour, general heritable influences and social deprivation virtually obliterated the association between PCSE and conduct disorders.17 Nonetheless, such negative findings do not rule out the contribution of an interaction between a specific gene(s) (not properly represented in the study’s cohort), environment (smoke exposures) and development. Indeed, just as cannabis use in adolescence appears to modulate the risk for the later development of schizophreniform symptoms only in those with a specific genetic vulnerability (i.e., carriers of Val allele at the catechol-O-methyltransferase [COMT] locus),25 it is conceivable, according to the cumulative data, that PCSE may add significantly to the risk of later developing conduct disorders only in carriers of a specific genetic vulnerability, such as the low monoamine oxidase A (MAOA) expressing allele.
The complex neurobiology of conduct disorders
Neural substrates of antisocial behaviours
Although the neurobiological basis of antisocial behaviour is poorly understood, most authors agree that the symptoms associated with conduct disorder (Fig. 1A) arise from a peculiar response to emotional cues in the social environment. The underlying deficiency appears to comprise 2 related but dissociable components: antisocial attitudes and impaired behavioural inhibition, or impulsivity. Proper behavioural responses to emotionally charged stimuli rely on a circuit that involves the amygdala,29 dorsolateral prefrontal cortex, the anterior cingulate gyrus30,31 and orbitofrontal cortex32 (Fig. 1B). An impaired ability to rein in aggressive behaviours is thought to emanate from lower fear response and higher anger reactivity (amygdala) combined with impaired inhibitory control by prefrontal regions (including the prefrontal cortex, orbitofrontal cortex and anterior cingulate gyrus).

Top–down view of conduct disorders. (A) Conduct disorders according to DSM-IV criteria.26 Conduct disorders can be suspected when a child displays a repetitive and persistent pattern of serious aggressive or nonaggressive misbehaviours against people, animals or property that may be characterized as belligerent, destructive, threatening, physically cruel, deceitful, disobedient or dishonest. A diagnosis of conduct disorder can be established by the presence of 3 (or more) of the included criteria in the past 12 months, with at least 1 criterion present in the past 6 months and with the disturbance in behaviour causing clinically significant impairment in social, academic or occupational functioning. (B) Key brain structures in the circuitry that controls emotion: (i) OPFC in green and the VMPFC in red, (ii) DLPFC, (iii) amygdala and (iv) ACG. Each of these interconnected structures plays a role in different aspects of emotion regulation, and abnormalities in one or more of these regions or in the interconnections among them are associated with failures of emotion regulation and also increased propensity for impulsive aggression and violence. (Davidson RJ, Putnam KM, Larson CL. Dysfunction in the neural circuitry of emotion regulation–a possible prelude to violence. Science 2000;289:591–4.27 Reprinted with permission from AAAS.) (C) Schematic model of one way in which developmental 5-HT disturbances may affect the neural circuitry regulating emotion, largely based on some of the reported effects of 5-HT transporter polymorphisms on the electrophysiology of the circuit. (Hariri AR, Holmes A. Genetics of emotional regulation: the role of the serotonin transporter in neural function. Trends Cogn Sci 2006;10:182–91.28 ©2006, with permission from Elsevier.) 5-HT = serotonin; ACG = anterior cingulate gyrus; DLPFC = dorsolateral prefrontal cortex; OPFC = orbital prefrontal cortex; PFC = prefrontal cortex; VMPFC = ventromedial prefrontal cortex.
Early animal studies had suggested that cortical projections to the amygdala are critical in assigning emotional significance to stimuli.33 It has also been observed that lesions to the orbitofrontal cortex and adjacent prefrontal cortex can lead to impulsivity and aggression.34 Human brain-imaging studies also support the notion that aggressive behaviours could be partly attributed to a disrupted amygdalar-frontal circuit.35–38 For example, a study of 40 adolescents who exhibited aggressive behaviours found decreased activity in dorsolateral prefrontal cortex and increased activity in the anteromedial portions of the frontal lobes, as well as activity changes in the limbic system.39 Similarly, compared with control subjects, persons with a diagnosis of conduct disorders have been reported to show a pronounced deactivation of the right dorsal anterior cingulate gyrus while experiencing negative affect-laden pictures,40 reduced right temporal lobe and right temporal grey matter volumes41 and a distinct electrophysiological defect in the frontal brain.42 Although not yet conclusive, the neuroimaging data help to delineate the brain areas associated with impulsive acts that may be dysfunctional in people with antisocial or aggressive behaviours.
The serotonergic system has been the most extensively scrutinized for its role in the development of such circuits.28 The resulting literature is vast and complex, partly owing to the fact that serotonin (5-HT) and other monoaminergic neurotransmitters also act as unconventional trophic factors, playing key roles during brain morphogenesis (see Nicotra and colleagues43 for a comprehensive review). Such complexity notwithstanding, the combined evidence appears to suggest that conduct disorders might be more strongly associated with decreases in 5-HT.44
In this context, we should pay particular attention to the amygdala, which, with its heavy serotonergic input,45 is likely to be uniquely sensitive to alterations in serotonergic neurotransmission. Serotonin appears likely to regulate the normal balance between excitatory and inhibitory neurotransmission, thus altering signal processing within the amygdala and modifying behavioural responses dependent on this processing44 (Fig. 1C). For example, selective serotonin reuptake inhibitor (SSRI)–mediated 5-HT increases have been reported to potentiate amygdala response to threat-relevant stimuli46 perhaps by disrupting the neural processes involved in the handling of social information.47 This mechanism could explain how genetically driven excesses in 5-HT during development might result in a lopsided prefrontocortical-amygdalar circuit (Fig. 1C) that compromises the function of neural circuits regulating emotion, negative affect and stress later in life.28,48
It seems particularly relevant to mention in this context the known 5-HT autoregulatory negative feedback, likely mediated by the 5-HT1A receptor, that ensures an appropriate level of serotonin innervation in the adult brain.49 Thus any behavioural effects of aberrant serotonergic transmission would be highly specific to developmental stage and could explain how an excess of 5-HT during fetal development could lead to decreased 5-HT in the adult brain, facilitating impulsivity and aggressive behaviour.50,51 Indeed, aggressive men may be more prone to aggression following reductions in plasma tryptophan,52 SSRIs reduce aggression in psychiatric patients, 53 and 5-HT has been consistently found to exert inhibitory control over impulsive aggression (see Davidson and colleagues27 and van Goozen and colleagues53 for reviews).
However, not all studies have shown these associations. For example, while lower concentrations of 5-HIAA, a 5-HT metabolite, have been reported to predict aggressive behaviours in boys with conduct disorders,54 findings of 5-HT increases and decreases, and of no differences, have also been reported.55 Such ambiguity in the combined data may reflect 5-HT’s exquisitely orchestrated developmental trajectories as well as the contribution of other factors (e.g., genetic, environmental and other monoaminergic systems) that may modulate the direction and magnitude of 5-HT’s roles (neurotransmission and morphogenesis) as well as the specificity of serotonin effects as a function of the brain region or circuit it modulates.
The stress response system is likely to be another major confounding factor and a significant contributor to individual differences in antisocial behaviour. Indeed, aggressive individuals appear to be less sensitive to stress, a phenomenon that has been proposed to result from either a relatively lower capacity to mount a fear response56 or a proclivity to engage in risk-taking behaviours.57 Regardless of the underlying mechanisms, there is consistent evidence of an inverse relation between the ability to increase cortisol in response to stress and the manifestation of antisocial behaviours.44 In the context of the stepwise model of conduct disorders being developed in this article (see below), it is worth mentioning a recently published hypothesis positing that an adverse childhood environment is likely to present frequent stressful stimuli, resulting in habituation and less reactivity to stress.44
In spite of the challenge presented by such confounds, aptly reviewed elsewhere,59 it is clear that 5-HT activity can contribute to the kinds of bias in information processing that could predispose a person to pathological conditions such as aggressive or antisocial behaviours. If, as we hypothesize here, there is inhibition secondary to maternal smoking of fetal brain MAO, which regulates monoamine levels, it follows that this could affect the integrity of the underlying networks.
Direct and indirect effects of nicotine on brain development and behaviour
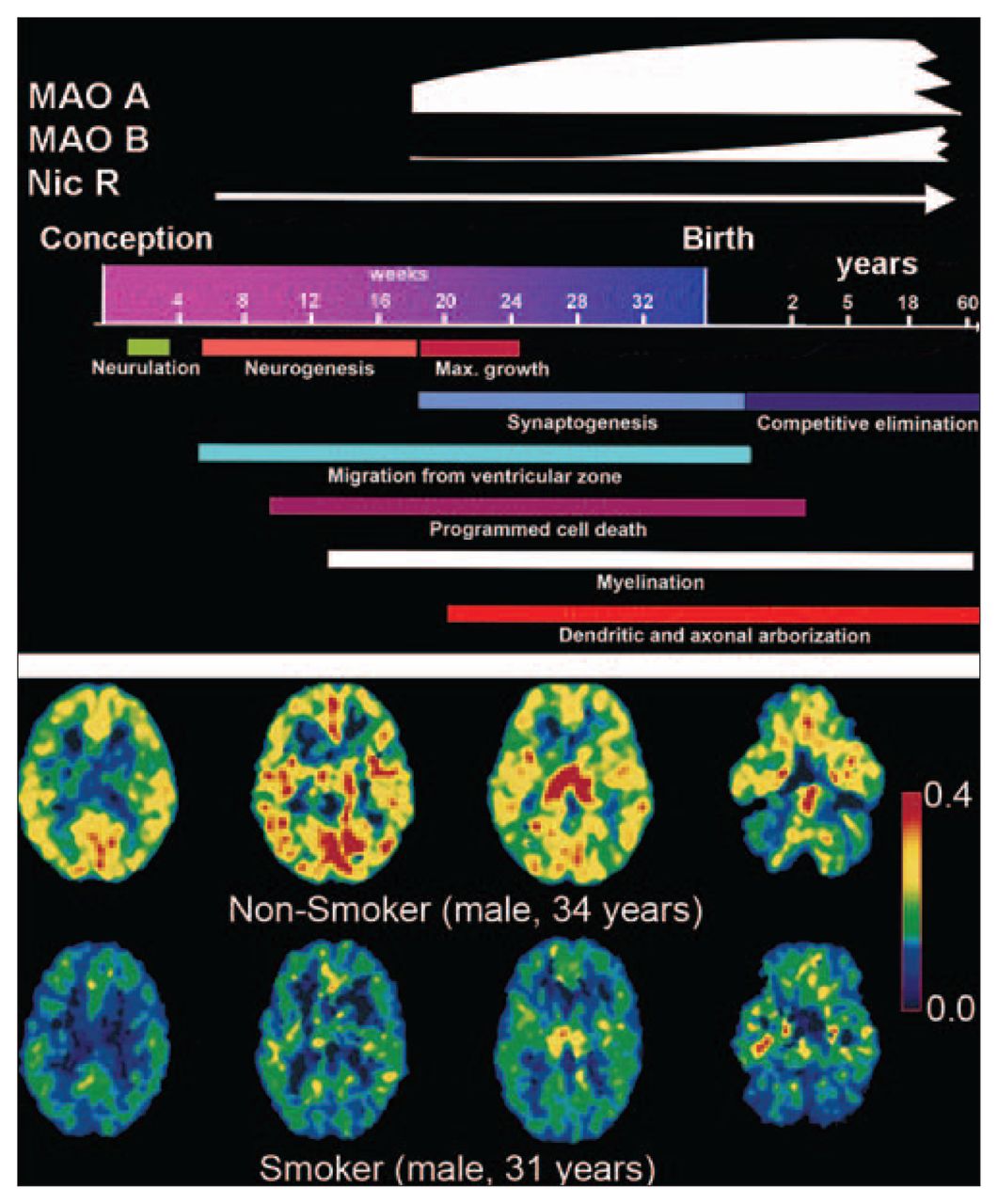
In tobacco smoke, there are thousands of toxic chemicals with the potential to derail key developmental processes either directly or indirectly, and nicotine is particularly relevant among them. Even though, or perhaps because, nicotinic acetylcholine receptors display many different properties and are ubiquitously expressed in the brain, their actual roles in normal brain physiology have been difficult to elucidate. Nevertheless, multiple lines of evidence suggest a significant and multifaceted role during brain development. Brain nicotinic receptors, which appear early60 and reach high levels during human gestation (Fig. 2, top panel), have been shown to affect gene expression, neuronal differentiation, synapse formation and neuronal path finding. Predictably, administration of nicotine to pregnant rats changes basal neuronal activity in fetal mesocorticolimbic structures,63 induces damage to serotonergic projections in the cerebral cortex and striatum, 64 limits growth of the forebrain and reduces the number of Purkinje cells in the cerebellar vermis,65 and compromises brain repair mechanisms.66

The rise and fall of MAO. (Top panel) Time course of nicotinic receptors and MAO’s expression is overlaid atop that of critical events in the determination of brain morphometry during (and after) human gestation. (From: Lenroot RK, Giedd JN. Brain development in children and adolescents: insights from anatomical magnetic resonance imaging. Neurosci Biobehav Rev 2006;30:718–29.61 ©2006, with permission from Elsevier.) (Bottom panel) Positron emission tomography images of the MAOA radiotracer [11C] clorgyline, the trapping of which is a function of both the concentration of MAOA and blood flow, as acquired at equivalent planes in the brains of a nonsmoker (top row) and a smoker (bottom row). The colour scale represents MAOA concentration (values of λk3 range from 0.4 [red] to 0 [black]). (From: Fowler JS, Volkow ND, Wang GJ, et al. Brain monoamine oxidase A inhibition in cigarette smokers. Proc Natl Acad Sci U S A 1996;93:14065–9.62 ©1996 With permission, National Academy of Sciences, USA.) MAO = monoamine oxidase.
Nicotine can also upregulate nicotinic receptors,67 which could perturb brain maturation. For example, endogenous nicotinic activity acts throughout the nervous system to switch GABAergic signalling from excitatory (during early stages of development) to inhibitory. Since nicotinic blockade can extend the period of GABAergic excitation,68 intrauterine exposure to nicotine could disrupt (delay) the timing of the switch, affecting the development of neurons and their proper integration into circuits.69 Interestingly, this may affect the broader serotonergic system because GABAergic axons traverse the developing raphe nuclei in the brainstem at a time when 5-HT neurons are just beginning to differentiate and migrate away from the ventricular zone.70 At the system level, nicotine also interacts with specific receptors in placental vasculature, disrupting the flow of blood and fluids and depressing active amino acid uptake and delivery.71 The resulting fetal undernutrition helps explain the underdevelopment of both neural and nonneural elements,72 which constitute recognized risks for internalizing and externalizing psychiatric disorders and for behavioural problems.73
Predictably, animal studies suggest several ways in which nicotine in the fetal brain could affect neurobehaviour and social development.19,74 For example, nicotine exposure during early embryonic stages abrogates the ability of 7- to 11-day-old chicks to learn a detour task, an effect associated with failure to upregulate ornithine decarboxylase activity75; as well, brain aromatase activity, known to play a role in sexual brain differentiation, is inhibited in male but not female offspring of pregnant rats infused with nicotine.76 Also, fetal nicotine exposure in rats affects the physiology of neurons in the pedunculopontine nucleus,77 a site that controls arousal, producing a more excitable offspring78; further, it alters 5-HT synaptic communication, changing the response to nicotine administration and conferring vulnerability to nicotine dependence later in adolescence.64 Taken together, these observations point to just a few of the possible complex mechanisms linking nicotine exposure to negative effects on behavioural substrates. More research is needed, however, to fully understand how these substrates are affected by the primary and secondary effects of exposure to nicotine in utero, as well as the possible (and likely) contribution of other neurotoxic components present in tobacco smoke.79
MAO and behavioural outcomes: a complex relation
In light of 5-HT’s critical role in guiding brain developmental processes, it is highly pertinent that some of the more than 4000 nonnicotinic components present in tobacco smoke,80 such as 2,3,6-trimethyl-1,4-naphthoquinone and farnesyl-acetone,81 the aromatic β carbolines, norharman and harman,82 and even carbon monoxide,83 are all suspected inhibitors of MAO enzymes, which catalyze the oxidative deamination of neurotransmitters and biogenic amines (e.g., dopamine, norepinephrine, tyramine and serotonin). This may account for tobacco smoke’s ability to reduce MAO activity in vitro,84 and in the periphery in vivo,85–88 and in a smoker’s brain and body62,87,89,90 (Fig. 2, bottom panel). Such whole-body effects strongly suggest that the MAO inhibitory activity could reach the brain of an exposed fetus, a phenomenon that has not yet been tested; if exerted during critical times of synaptogenesis (Fig. 2), this inhibitory activity could conceivably potentiate the nicotinic damage to 5-HT–dependent morphogenetic processes.
The time-sensitive impact of alterations in morphogenetic neurotransmitters
When considering the possibility that smoke-driven inhibition of MAO also occurs in fetal tissues, it is worth noting that relatively high concentrations of MAOA enzyme activity appear significantly earlier in the developing human brain, compared with MAOB,91 which begins its rise toward adult levels perinatally92 (Fig. 2). This could be significant since MAOA (but not MAOB93) gene knockout (KO) mice display increased aggressiveness when compared with wild-type littermates. 94 Also, pharmacologic inhibition of MAOA (and B) during fetal development, but not during adulthood, results in aggressive phenotypes in rodents.95,96
A depressed MAOA activity could boost the nicotine-mediated surges in serotonergic, noradrenergic and dopaminergic tone.97 Indeed, significantly higher levels of 5-HT can be found in the ventral hippocampus (+202%), frontal cortex (+96%) and dorsal raphe nucleus (+147%) of MAOA KO mice.98 In turn, tonic increases in 5-HT may contribute to some of the developmental disruptions observed in the brains of these mice, such as aberrant 5-HT projections99 and the malformations in the barrel field of the primary somatosensory cortex100 in serotonin transporter gene KO mice.
These neurochemical deficits foreshadow a growing neurobehavioural literature suggesting that early interference with the MAO system could enhance the risk of antisocial or aggressive behaviours, or both. Consistent with this notion, in preclinical94–96 and clinical101–103 studies, fetal reductions in MAOA have been linked to a range of behavioural effects related to aggression or impulsivity in the offspring. Because of the monoamines’ dual role in neurotransmission and morphogenesis, however, there is no reason to expect that the behavioural outcomes of pharmacologic 5-HT manipulations during adulthood should recapitulate the effects of 5-HT disruptions during brain development.
The genetics of MAOA and conduct disorders
There is a well-established genetic contribution to the risk for conduct disorders, with both aggressive and nonaggressive forms of antisocial behaviours displaying measurable (sometimes as high as 60% of observed covariance) heritable components.104–106
Predictably, MAOs have been a major focus of research attempting to map the genetic pathways leading to conduct disorders. Interestingly, the fact that MAOs are X-linked genes may explain, at least in part, the significantly higher prevalence of conduct disorders among boys, since males carry only 1 allele that determines expression levels.
With regard to other potentially relevant genetic differences, much attention has been devoted to MAOA regulatory variants associated with either low or high levels of gene expression. The polymorphism most commonly scrutinized in this context is a variable number of tandem repeats (VNTR) in the promoter of the MAOA gene that engenders an allelic imbalance,107 so that a 4-repeat VNTR confers higher levels of reporter gene expression than a 3-repeat VNTR.108
There is growing albeit inconclusive evidence that functional variations in the MAOA gene, as well as other gene products that modulate serotonergic tone,109 modulate the risk for conduct disorders. Expectedly, and mirroring the bidirectional findings at the level of 5-HT, the issue of whether the high- or the low-activity allele confers protection is still a matter of debate. For example, and in support of the former hypothesis, maltreated children appear to be at a higher risk for developing antisocial behaviours if they carry the low, as compared with the high, MAOA allele103; the former has also been linked to socioemotional hypersensitivity. 110 Also, a point (null) mutation in the structural MAOA gene has been identified in a large Dutch kindred, in which several affected male subjects (essentially human MAOA knockouts) suffered borderline mental retardation and displayed distinct abnormal behaviours including impulsive aggression, arson, attempted rape and exhibitionism.102 Conversely, associations have also been reported between the high MAOA allele and aggressiveness and impulsivity in both healthy men111 and in patients with ADHD.112 Finally, a recent brain-imaging study showed that carriers of the high-activity allele scored worse on a task that measures impulsivity, compared with the carriers of the low-activity allele, in a way that correlated with greater blood oxygen level dependent responses in several cortical areas.113
The combined evidence suggests that MAOA polymorphisms may modulate aggressive behaviour in humans. The connections to conduct disorders, however, are likely to be complex and indirect as they influence developmental processes and their interaction with environmental factors. In one study, for example, an MAOA polymorphism was found to be predictive of antisocial behaviour only in those subjects who were exposed to physical abuse during childhood.103 This observation is consistent with the results of a study in nonhuman primates showing that the interaction between a low expressing MAOA allele and psychosocial stress correlated with higher levels of aggressive behaviours in the offspring.114
PCSE and MAO: effects at the confluence of genetics, brain development and early environmental factors
Here we propose that the contribution of MAOA deficiencies toward conduct disorders are most likely restricted to an early window of vulnerability in the process of brain development. Indeed, using imaging we found no differences in MAOA levels in brain as a function of MAOA genotype in healthy adult subjects.115 These results are similar to those obtained with cortical autopsy samples116 and convergent with previous examples in which the expected influence of genotype on protein expression in the adult brain was inconsistent or very small.117 Thus the putative deleterious effects of carrying a low expressing MAOA allele or of inhibiting MAOA enzyme activity with tobacco smoke may derive from interactions with specific developmental factors, such as the lack of sufficient compensatory MAOB expression in the fetal brain during development, when the brain is undergoing major changes. Alternatively, different alleles could in theory display very different and developmentally segregated patterns of gene expression, a possibility worth investigating.
What brain developmental processes may be susceptible to the contingent variations in MAOA gene expression? A possible answer is hinted at by a study showing that a low expressing MAOA allele — the one previously linked in some studies to an increased risk of violent behaviour — was associated with smaller volumes in limbic structures (amygdala and anterior cingulate gyrus), larger volumes in lateral orbitofrontal cortex (only in male subjects) and concomitant increased amygdalar responsiveness and decreased reactivity in regulatory prefrontal regions58 (Fig. 3A and 3B).

Magnetic resonance imaging data demonstrate limbic and paralimbic regional volume changes in subjects with the low-activity MAOA variant (n = 97). Plots represent the summed volumes of voxels in predefined regions of interest, normalized to volume measures relative to the high-activity MAOA group mean (100%). (A) Compared with high MAOA subjects, low MAOA subjects exhibit significant volume reductions in bilateral amygdala, supragenual anterior cingulate and subgenual anterior cingulate cortex. Male and female subjects were combined. (B) Male low MAOA subjects showed increased lateral orbitofrontal volume, bilaterally, relative to high MAOA subjects. (From: Meyer-Lindenberg A, Buckholtz JW, Kolachana B, et al. Neural mechanisms of genetic risk for impulsivity and violence in humans. Proc Natl Acad Sci U S A 2006;103:6269–74.58 ©2006 With permisssion National Academy of Sciences, USA.) MAOA-H = high expressing monoamine oxidase-A variant; SEM = standard error of the means.
The differential impact of MAOA alleles on the morphology and function of brain regions implicated in cognitive control and the generation and regulation of emotions could explain at least part of the previously reported association between MAOA, impulsivity and aggression. Serotonin levels, which are especially sensitive to MAOA activity, appear to play a major regulatory role during neurodevelopment,118,119 particularly in the formation of a properly counterbalanced cortico-amygdalar circuit. Hence, the morphologic correlates of low MAOA levels would indicate that agents affecting MAOA early in development have the potential to affect significant long-term neurologic changes poised to hamper the emergence of normative social behaviours. Importantly, since the subjects in this study58 were psychiatrically normal, one must conclude that the observed MAOA-related structural and functional differences in corticolimbic circuitry must, by themselves, be compatible with normal mental health.
Summary and agenda for the future
Collectively, the findings described here offer a tantalizing example of how direct effects on the MAO system could modulate the complex interactions between genetic, developmental and environmental factors, as highlighted in recent models,53,120 to increase the risk of conduct disorders in the offspring. Further research on such complex interactions may provide better tools with which to assess the ambiguous nature of the available epidemiologic evidence of increased risk for conduct disorders in the offspring of mothers who smoked while pregnant.
From the evidence we have synthesized here, we propose a modified hypothetical model (Fig. 4), according to which smoking-induced MAO inhibition, when combined with allelic variations that lower MAOA gene expression, place the offspring, via the modulation of brain development trajectories, at a higher risk for developing conduct disorders later in life.

Hypothetical model of the cumulative effect of various risk factors for developing conduct disorder. Normal brain development (A arrow) relies, among other factors, on carefully balanced concentrations of monoamine neurotransmitters in time and space. Genotypes that reduce MAOA activity during fetal development (B arrow) apply downward pressure on this process, particularly in males, who have only 1 allele. The magnitude of this pressure can induce morphologic changes in limbic and cognitive areas (i.e., significant volume reductions in the cingulate gyrus and bilateral amygdala [blue areas] as well as volume increases in the lateral orbitofrontal cortex [red spots] only in males)58 that, by themselves, seem compatible with behavioural outcomes within the normal range. Prenatal exposure to gonadal sex steroids (C arrow) is a powerful driver of differential neural circuit formation in the developing brain and no doubt a contributor to the higher aggressive and impulsive tendencies normally observed in males. Because of the neurodevelopmental impact of stress mechanisms, adverse (stressful or traumatic) childhood environments (D arrow) can push vulnerable children over the threshold and significantly increase the chances for developing disruptive behaviours later in life, even though genotypic differences in MAO enzymes no longer translate into different levels of MAO expression in adults. Many investigators have suggested that the risk for conduct disorders could become exacerbated by intrauterine exposure to tobacco smoke (E arrow) because nicotine usurps receptors that are critical during normal brain development and because other components in tobacco smoke can affect the development of circuits involved in the control of emotions through further reductions in MAO activity in the fetus. For the sake of simplicity, the genetically (MAOA high) resilient brain represented at the top is largely unaffected by the various downward vectors, but in reality, many combinations of heavy exposure to adverse environmental conditions and different genetic vulnerabilities can similarly lead to pathological behavioural outcomes. MAOA = monoamine oxidase type A.
The developmental processes modulated by MAO (but also by the dopamine transporter, COMT and sex specifiers) at these stages constitute a target for many converging insults, and the risk may step over a threshold when a stressful or adverse environment is added to the equation. This hypothetical model now awaits the research needed to corroborate that smoking inhibits MAO in the fetal brain and that prenatal exposure to cigarette smoke confers a higher risk for conduct disorders in those infants carrying the low MAOA allele.
Footnotes
Medical subject headings: monoamine oxidase; smoking; maternal-fetal exchange; conduct disorder.
Competing interests: None declared
Contributors: Drs. Baler and Volkow designed the review and wrote the article. Drs. Volkow and Fowler acquired the data, which Drs. Baler, Volkow and Benveniste interpreted. All authors reviewed the article and gave final approval for its publication.
- Received August 30, 2007.
- Revision received October 23, 2007.
- Accepted November 12, 2007.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.