

Abstract
The goal of my research is to gain insight using rodent models into the fundamental molecular, cellular and systems that make up the base of memory formation. My work focuses on fear memories. Aberrant fear and/or anxiety may be at the heart of many psychiatric disorders. In this article, I review the results of my research group; these results show that particular neurons in the lateral amygdala, a brain region important for fear, are specifically involved in particular fear memories. We started by showing that the transcription factor CREB (cAMP/Ca2+ response element binding protein) plays a key role in the formation of fear memories. Next, we used viral vectors to overexpress CREB in a subset of lateral amygdala neurons. This not only facilitated fear memory formation but also “drove” the memory into the neurons with relatively increased CREB function. Finally, we showed that selective ablation of the neurons overexpressing CREB in the lateral amygdala selectively erased the fear memory. These findings are the first to show disruption of a specific memory by disrupting select neurons within a distributed network.
A fundamental goal of neuroscience is to understand how memories are encoded and stored in the brain. Ensembles (or groups) of neurons are thought to serve as the physical representation of memory (the memory trace or “engram,” a term first coined by Richard Semon in 19211). However, identifying the precise neurons that constitute a memory trace has been challenging neuroscientists since Karl Lashley began his famous search for the elusive engram in the 1920s. In his studies, Lashley trained rats in various tasks (such as traversing a maze to find a food reward) and lesioned various portions of different cortical regions either before or after training. After 30 years of work, he summarized his findings in a seminal paper entitled “In search of the engram.”2
Lashley observed that cortical lesions disrupted performance in the maze and that the degree of disruption was roughly proportional to the amount of cortex removed but not to the location of the lesion. From this, he concluded that all cortical areas can substitute for each other as far as learning is concerned (the principle of equipotentiality) and that the cortex tends to act as a whole in that the amount, rather than the location, of cortical tissue removed correlated with performance in the maze (the principle of mass action). These findings led Lashley to conclude that memories are not localized, but rather distributed, in the cortex. In his words,
“…this series of experiments has yielded a good bit of information about what and where the memory trace is not. It has discovered nothing directly of the real nature of the memory trace. I sometimes feel, in reviewing the evidence of the localization of the memory trace, that the necessary conclusion is that learning is just not possible. It is difficult to conceive of a mechanism that can satisfy the conditions set for it. Nevertheless, in spite of such evidence against it, learning sometimes does occur.”2
Lashley’s failure to find the engram may have been because of his choice of memory task (learning a maze is a complex task, and this sort of memory likely relies on many brain regions) and brain region examined (regions other than the cortex may be involved in memory).
Since Lashley’s studies, though, there has been progress in “finding the engram.” Both cellular imaging and electro-physioloic studies3–7 have detected neurons whose activity is correlated with memory encoding or expression or both, suggesting that these active neurons make up the memory trace. For example, John Guzowski’s laboratory used a technique they developed, referred to as catFISH (compartmental analysis of temporal activity by fluorescence in situ hybridization),8 and found that some of the same neurons in the dorsal hippocampus of rats were active when the rats were re-exposed to a unique environment. More recently, Mark Mayford’s laboratory used a sophisticated transgenic mouse approach that allowed them to tag active neurons.5 They showed that some of the same amygdala neurons that were active during fear learning were also active during the retrieval of that fear memory. Furthermore, the number of reactivated neurons in the amygdala was correlated with the strength of the retrieved fear memory.
These findings suggest that specific neurons are involved in a memory. However, these correlative studies do not address whether these active neurons are essential components of the memory trace. A direct test of this hypothesis would require specifically disrupting only these activated neurons while leaving their neighbours intact and then determining whether subsequent memory expression is blocked. Establishing such a causal role for particular neurons in a memory has been difficult because the neuronal ensembles that make up this memory trace are thought to be sparsely distributed,9 and the ability of current pharmacological, genetic and lesioning techniques to target specific subsets of neurons within a brain region is limited.
In my laboratory’s “search for the engram,” we used auditory fear conditioning as our memory task. In this Pavlovian paradigm, a neutral tone is paired with a mildly aversive footshock. One tone-shock pairing is sufficient to produce a robust and long-lasting fear memory that can be quantified by measuring the percentage of time mice spend freezing (an adaptive defensive response) when the tone is subsequently replayed. Automated measures have been established to easily and reliably quantify freezing.10 We chose this task because genetic, biochemical, electrophysiological and behavioural studies from many laboratories, including those of Mike Davis, Michael Fanselow, Joe LeDoux and Steven Maren, established that lateral amygdala is required for auditory-conditioned fear memories11–15 (but see16). Now we knew where to look in the brain for a fear memory trace. The next step was to target lateral amygdala neurons that are active following fear training or testing.
To target neurons whose activity is correlated with memory, Jin-Hee Han, Adelaide Yiu and Hwai-Lin (Liz) Hsiang in my laboratory, together with our collaborators Steven Kushner, Bruno Bontempi and Paul Frankland, took advantage of our recent findings that increasing the level of the transcription factor CREB (cAMP/Ca2+ responsive element binding protein) in a small portion of lateral amygdala neurons enhances auditory fear memory under certain training conditions.17 Furthermore, we found that neurons with relatively increased CREB levels are more likely than their neighbours to be active following fear memory training or testing. This suggested to us that these neurons with high CREB levels outcompete their neighbours for inclusion in the fear memory trace.
The idea that CREB is involved in memory is not new. Indeed, studies by Tim Tully’s and Eric Kandel’s laboratories showed that CREB is necessary for memory formation in invertebrates.18–22 Parallel studies in rodents have also showed that CREB is important for memory formation23–34 (but see35).
I first began examining the effects of increasing CREB function on memory when I was a postdoctoral fellow at Yale in Mike Davis’s laboratory. Eric Nestler’s laboratory was located across the hall at the Connecticut Center for Mental Health, and Bill Carlezon, a postdoctoral fellow in his laboratory at the time, was manipulating CREB function using replication-defective herpes simplex viral (HSV) vectors to determine the effects of CREB on cocaine-induced drug seeking.36 Although there were many ways to decrease CREB function, there were relatively few ways to transiently enhance its function in rodents. The HSV system was an ideal tool to study the effects of enhancing CREB on memory. This system, which allows expression of a transgene in a particular brain region at a particular time, was largely pioneered by my colleague, Rachael Neve, now at the Massachusetts Institute of Technology.
Herpes simplex viral vectors offer many advantages over other viral vector systems.37 First, HSV infects adult (nondividing) neurons (rather than glia) with relatively high efficiency.38,39 Second, the DNA from HSV remains episomal, thus avoiding potentially confounding effects of integration into the host DNA. Third, these vectors offer a large capacity for the insertion of foreign DNA. In our studies, we drove transgene expression with the HSV immediate–early gene IE4/5 such that transgene expression peaked 2–5 days following infusion and declined within 7 days.40,41 In this way, we could examine the effects of acutely increasing CREB function on fear memory in the lateral amygdala. In collaboration with Bill Carlezon and Eric Nestler, Mike Davis and I showed that increasing CREB levels in the lateral amygdala enhanced memory for fear-potentiated startle, another fear memory task,12 in rats that had been given weak training (consisting of several training trials massed together without intervening rest periods, which has been shown to produce weak memory).42 What intrigued us about our finding was that we observed robust memory enhancement despite the fact that we increased CREB levels in a relatively small percentage of lateral amygdala neurons (our HSV vectors infected roughly 15% of lateral amygdala neurons). This disconnect between the small number of neurons infected but large behavioural results continued to motivate my experiments.
Later, when I was a postdoctoral fellow in Alcino Silva’s laboratory, Steven Kushner, Alcino and I had long conversations about how we might make sense of these findings. We definitely believed the data. Mike Davis taught me to always follow the data (to borrow from Shakira, data, like hips, don’t lie). Plus, our basic effect was replicated by 2 additional groups using the same HSV vectors.43,44 We reasoned that one way to explain the data was that the low number of neurons with increased levels of CREB were somehow outcompeting their neighbours for inclusion in the memory trace. When I began my laboratory in Toronto, Jin-Hee Han, a talented postdoctoral fellow in my laboratory, continued these studies.
First, we revamped our viral vectors to allow infected neurons to be easily visualized by fusing the transgenes with green fluorescent protein (GFP). Tagging the N-terminus of CREB with GFP does not interfere with the functional activity of CREB.37,40,45,46 In our first studies, we used 3 main vectors: HSV-CREB expressing wild-type CREB (CREBWT); HSV-mCREB (the mutant CREB vector) expressing a mutated version of CREB that cannot be phosphorylated at the key Ser133 residue (CREBS133A); and HSV-GFP (control, Cntrl vector) expressing GFP or LacZ.
Our overall aim was to determine whether neurons infected with the CREBWT vector were preferentially recruited to the lateral amygdala fear memory trace. We reasoned that if competition between neurons for inclusion in the memory trace occurred during learning, the easiest way to see this would be to “stack” the experimental deck by maximizing the difference between the neurons infected with the CREBWT vector and their noninfected neighbours. Thus, we first infused our CREBWT vector into the lateral amygdala of mice with a targeted disruption of the 2 main isoforms of CREB, CREBαδ−/− mice.47 The CRE–DNA binding is virtually abolished (by > 90%),48,49 and the levels of CREB protein are dramatically reduced (roughly 85%–90% reduction compared with controls) in the brains of CREBαδ−/− mice.48,50 These CREB-deficient mice have previously been shown to be impaired in auditory fear conditioning.23,28 Importantly, we replicated this memory deficit and showed that infusing our Cntrl vector into the lateral amygdala did not change this. However, infusing the CREBWT vector into the lateral amygdala of these mice completely rescued the auditory fear deficit; the CREB-deficient mice now froze at the same levels as their wild-type littermate control mice. Furthermore, under weak training conditions, we found that increasing CREB function in a similar small fraction of lateral amygdala neurons enhanced memory in wild-type mice (similar to my initial findings using fear-potentiated startle in rats42). Together these findings suggested that these neurons with increased CREB function were preferentially recruited to the trace supporting the fear memory.
We were inspired to explore this possibility further by John Guzowski’s elegant studies using the catFISH technique. Specifically, Jin-Hee Han, Adelaide Yiu and Christina (Christy) Cole in my laboratory, together with our collaborators, Steven Kushner, John Guzowski and Alcino Silva, attempted to visualize the fear memory trace by taking advantage of the unique transcriptional time course of the activity-dependent gene Arc (activity-regulated cytoskeleton-associated protein; Arg3.1).6,8 Under basal conditions, neurons contain very low levels of Arc RNA. However, neuronal activation (the type of activation that is associated with learning or long-term potentiation) produces a rapid but transient burst in Arc RNA synthesis (within about 3–5 min of activation).8 This RNA is then delivered to dendrites within roughly 20 minutes of activation. Therefore, the localization of Arc RNA (either in the nucleus, cytoplasm or both) can serve as an activity marker for that particular neuron, with Arc RNA localized to the nucleus being a molecular marker of a neuron that was active 5 minutes ago. We used this technique to identify neurons that were activated by fear memory training or testing and asked whether these neurons were also the ones we infected with the CREBWT vector.
We observed that neurons infected with the CREBWT vector were 3 times more likely than their noninfected neighbours to be Arc positive in wild-type mice and 10 times more likely in CREB-deficient mice. In contrast, neurons infected with the dominant-negative CREB vector were 12 times less likely than their neighbours to be Arc positive. This was consistent with our behavioural data showing that disrupting CREB function (using the CREBS133A vector) in roughly 20% of lateral amygdala neurons had no effect on memory in wild-type mice, perhaps because the remaining neurons (over 80%) with normal levels of CREB were sufficient for normal memory.17
Together with many control studies, these findings suggest that neurons with relatively higher CREB function are preferentially recruited to the memory trace. Would this mean that increasing CREB levels in all lateral amygdala neurons would enhance fear memory? We hypothesized that it would not, because we think that competition between eligible neurons is critical in memory enhancement. We reasoned that raising CREB levels in all neurons would not produce a stronger memory because all of the neurons would again be equal (and the signal-to-noise ratio would not be enhanced). As Gore Vidal put it, it is not enough to succeed; others must fail. The same may apply to neuronal competition underlying memory formation.
Our findings suggested that neurons with higher levels of CREB “win” the competition between neurons for inclusion in a memory trace. My laboratory then attempted to use this property to determine if selectively ablating just these neurons after training disrupts the expression of an established fear memory. If so, this would indicate that these neurons are critical components of the elusive memory trace. We used several techniques to do this but had no luck. It turns out that these neurons, , like Steven Segal, are hard to kill, perhaps because CREB is a survival factor.51 Then, Steven Kushner, an avid reader of scientific journals, came across a paper in Nature Methods by Ari Waisman and colleagues52 describing a novel transgenic mouse used for cell lineage ablation studies based on diphtheria toxin (DT). We decided to try to kill neurons overexpressing CREB using this approach.
First, a bit of background on DT. Corynebacterium diphtheriae produces DT, a potent toxin that, once it binds to its receptor (diphtheria toxin receptor; DTR) and is internalized into a mammalian cell, efficiently blocks protein synthesis to cause rapid apoptotic cell death.53–55 For cell death to occur using the DT-based system, both key components (DT and DTR) are required. Interestingly, mice do not normally express a functional DTR.56–58 So, Waisman and colleagues52 engineered a mouse that expresses a simian DTR transgene (driven by the ubiquitous Rosa promoter; Fig. 1). However, expression of DTR is dependent on the cre recombinase-mediated removal of a transcriptional STOP cassette (upstream of the DTR transgene they placed a floxed STOP cassette that silences DTR expression until the STOP cassette is removed by cre recombinase-mediated recombination). Systemic injection of DT anytime thereafter induced apoptosis, but only in the cells that have undergone cre-mediated recombination and express DTR. A single internalized catalytically active fragment of DT (DT-A) is sufficient to kill a cell, indicating the sensitivity of DT-induced cell ablation.59 Importantly, neither high doses of DT in wild-type mice nor the expression of DTR alone (without DT) induces apoptosis, indicating the specificity of the system.60,61 Because DT readily crosses the blood–brain barrier,62 these inducible DTR (iDTR) mice have been used to induce cell death in the brain.52,60

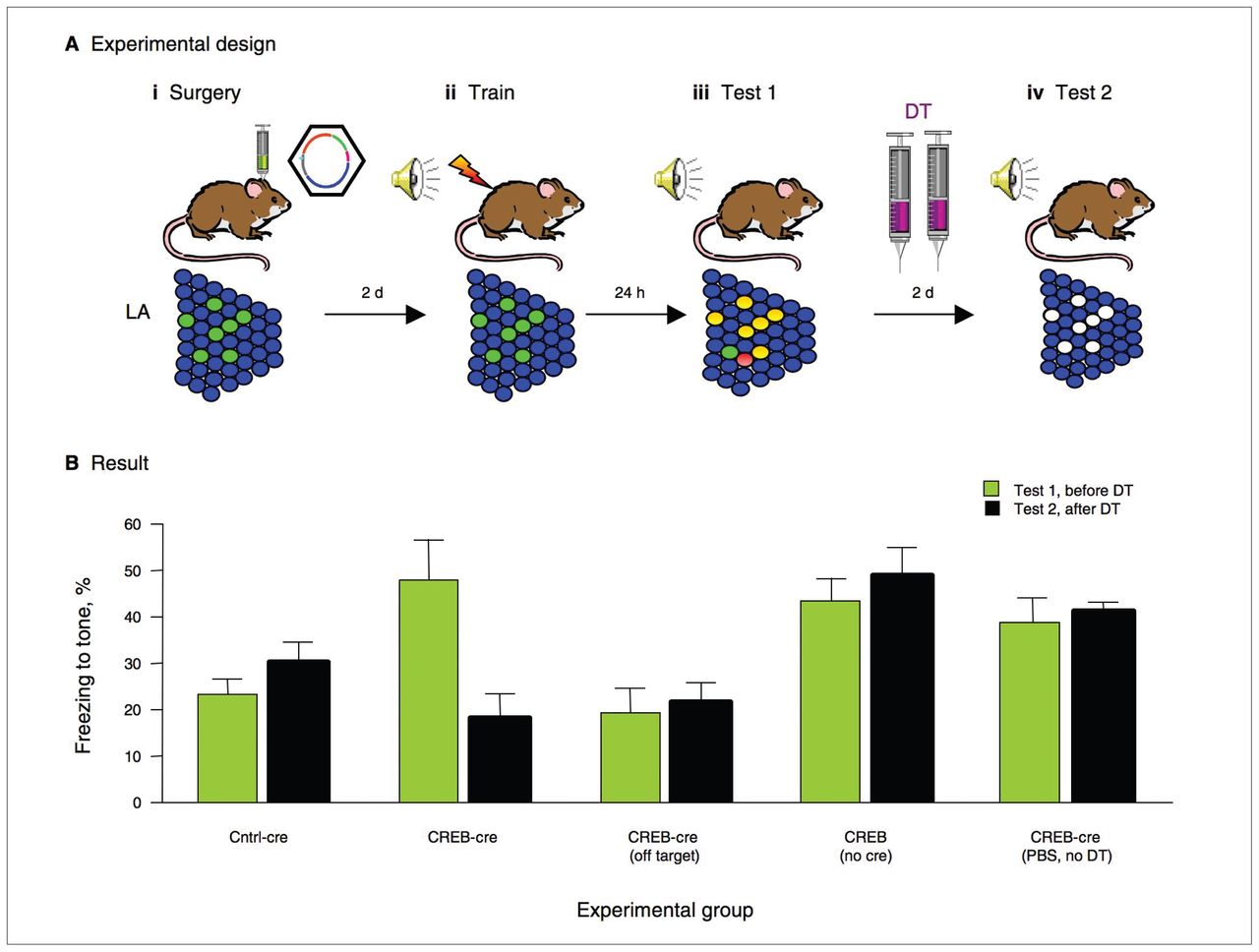
Schematic diagram depicting the strategy for selectively ablating neurons using inducible diphtheria toxin receptor (iDTR) transgenic mice. The mice express a simian DTR transgene. Expression of the DTR transgene is suppressed by a STOP cassette, which is floxed and removed when a neuron is infected with herpes simplex virus expressing cre recombinase (cAMP/Ca2+ response element binding protein [CREB]–cre vector or control-cre vector). Only neurons that have undergone cre-induced recombination will constitutively express DTR on the cell surface. At any point thereafter, systemic injection of diphtheria toxin (DT) induces apoptosis only in cells expressing DTR. In this way, only neurons infected with CREB-cre vector or Cntrl-cre vector will be ablated.
Many strategies can be used to delete defined cells, including the expression of cytotoxic proteins,63 antibodies64 or chemicals.65 However, these systems, and indeed earlier versions of the DT system, were not suited for our studies because they lacked temporal control over cell death and/or are prone to “leakiness” (in that cell death is not temporally or spatially limited to the targeted cell population).63 The iDTR mice were designed to address these drawbacks. Because DT induces cell death by apoptosis, it is thought to produce less inflammation than necrosis-induced cell death.66 In this way, the iDTR mice were designed to minimize the “bystander effect,” in which the death of a targeted cell directly or indirectly affects neighbouring neurons.
Jin-Hee Han, together with other members of my laboratory and our collaborators Steven Kushner and Paul Frankland, took advantage of the iDTR transgenic mice to selectively induce cell death in the neurons that we hypothesized were preferentially involved in the memory trace (neurons infected with the CREBWT vector). To this end, we re-engineered our viral vectors to include cDNA for cre recombinase. Our first vector expressed both CREB and cre recombinase (CREB-cre vector). When injected into the lateral amygdala of the iDTR mouse, we expected that a small portion of neurons would have increased levels of CREB and that a subsequent injection of DT would ablate just these neurons. Recombination and DTR expression can only occur in neurons expressing cre recombinase, allowing us to persistently tag infected neurons for subsequent ablation. This tagging is critical because of the relatively brief time course of transgene expression using HSV.40 Because this technique produces a small lesion of the lateral amygdala that may affect memory on its own, we needed a control vector. So we added cre recombinase to our Cntrl vector. Our thinking was that this Cntrl-cre vector would ablate a similar number of lateral amygdala neurons but that these neurons would be randomly located in the lateral amygdala and not involved in the memory trace (because they did not overexpress CREB). As a first step in this study, Jin-Hee confirmed that this iDTR/vector system selectively induced cell death by examining 2 markers of apoptosis (activated caspase 3 and terminal deoxynucleotidyl transferase dUTP nick end labeling [TUNEL]). We quantified the number of cells with activated caspase 3 and those that were positive for TUNEL and found that significant apoptotic cell death was only observed in the experimental groups (iDTR mice infused with either the CREB-cre or Cntrl-cre vectors). Importantly, we found that the CREB-cre and Cntrl-cre vectors produced a similar level of cell death. We were excited because we thought that this system, which allowed us to temporally ablate tagged neurons, would bring us one step closer to finding the engram.
To examine the effects of post-training ablation of neurons overexpressing CREB, we performed a series of behavioural tests. First, we found that increasing CREB levels in a population of lateral amygdala neurons (by microinjecting the CREB-cre vector) enhanced memory following weak training in iDTR mice. This finding replicated our earlier finding using the CREBWT vector (without cre recombinase) in wild-type mice.17 This memory enhancement, however, was completely reversed after systemic administration of DT (to delete just those neurons infected with the CREB-cre vector; Fig. 2). Importantly, the reversal of this memory enhancement was not observed in the control groups that lacked a key component of the DT-killing system (either cre recombinase or DT), consistent with the lack of cell death in these control groups. This was important because it showed that the memory reversal was not due to fear memory extinction caused by the second memory test.

Neurons with relatively increased CREB (cAMP/Ca2+ response element binding protein) function are essential for memory recall. (A) Schematic diagram of an experiment designed to test whether neurons with increased CREB function at the time of training are required for subsequent memory recall. (i) A subset of lateral amygdale (LA) neurons are infected with a CREB-cre vector (labelled green via green fluorescent protein [GFP]), which increases CREB in these neurons and induces them to undergo cre-induced recombination to express diphtheria toxin receptor (DTR). (ii) Mice are trained. A subset of LA neurons have high CREB levels, but because diphtheria toxin (DT) has not been administered, there is no cell death. (iii) Mice are tested. Neurons involved in the memory trace are depicted in red. Neurons with increased CREB out-compete their neighbours for inclusion in the fear memory trace (labelled yellow [green+red]). (iv) Mice receive systemic injections of DT to induce cell death only in the cells that express DTR (have undergone cre-induced recombination and, therefore, must have been infected with the CREB-cre vector). Mice are tested again. Neurons that previously had high CREB (infected) are now ablated. (B) Results from this experiment indicate that selectively ablating neurons with increased CREB at the time of training impairs subsequent memory recall. The CREB-cre group shows that increasing CREB function in a portion of neurons at the time of training enhances memory (Test 1) and subsequent ablation of these particular neurons completely reverses this enhancement (Test 2). The Cntrl-cre group show that GFP does not enhance memory and that selectively ablating these cells does not change this. The off-target group shows that increasing CREB function in neurons outside the LA does not enhance memory. The CREB alone (no cre) group shows that increasing CREB in the LA enhances memory (Test 1) and that DT injection (with no recombination) does not reverse this enhancement Test 2). The mice that received phosphate-buffered saline but no DT (PBS, no DT) show that CREB enhances memory in inducible DTR mice, and a systemic injection of PBS (rather than DT) does not change this on the second test day.
Next, we showed that deleting neurons infected with the CREB-cre vector (but not a similar portion of random neurons infected with the Cntrl-cre vector) also impaired the expression of a strong memory. Therefore, even though increasing CREB function in a portion of lateral amygdala neurons did not further enhance memory in mice that were trained using a strong protocol, these neurons are nevertheless important in the fear memory. In both of these experiments, however, we administered DT after a fear memory test. This memory test, however, may reactivate the fear memory and trigger a second wave of consolidation (referred to as reconsolidation).67 Karim Nader, Joe LeDoux and several other groups have shown that, similar to initial consolidation, reconsolidation requires protein synthesis.67 Because DT disrupts protein synthesis, we wanted to examine whether the memory disruption that we observed in the previous experiments was due to a disruption in reconsolidation. Therefore, we examined additional groups of mice but did not reactivate the memory (we did not give the mice a memory test) before DT administration. Instead, we administered DT at the same time but in the homecage. Again, we observed that mice microinjected with the CREB-cre vector and administered DT showed impaired memory whereas similarly treated Cntrl-cre mice did not.
If neurons overexpressing CREB during training (neurons with the CREB-cre vector) are critically involved in the memory trace, deleting this subpopulation should permanently block memory expression. To examine the persistence of memory loss, we trained mice using a strong protocol, administered DT in the homecage and assessed memory 2, 5 and 12 days later. The memory loss observed in the CREB-cre vector mice was long lasting; mice showed low freezing during the tone over these repeated tests. In contrast, memory remained robust over repeated tests in similarly treated Cntrl-cre mice. Therefore, we found no evidence of memory recovery in mice in which neurons overexpressing CREB were deleted, indicating that the memory was not just transiently suppressed.
To determine whether the memory loss could be due to a nonspecific impairment of lateral amygdala function (the mice did have a small lesion of the lateral amygdala), we retrained these mice. Following retraining, both groups of mice (CREB-cre and Cntrl-cre mice) showed equally high levels of freezing during the tone. That is, the post-training ablation of neurons that were overexpressing CREB (and cre recombinase) at the time of training produced a memory deficit, but these mice were capable of learning a tone-shock association (and freezing). Similarly, we also found that ablating neurons with the CREB-cre vector before fear conditioning did not impair subsequent memory formation. In this experiment, we infused the CREB-cre vector and systemically administered DT (to kill the neurons overexpressing CREB) before strong training and tested fear memory daily for 3 days. We found that even though a subpopulation of neurons was ablated before training, the mice acquired memory normally. Furthermore, this memory was neither more prone to extinction nor more fragile than a memory acquired without this killing (iDTR mice microinjected with the CREB-cre vector but systemically administered phosphate-buffered saline instead of DT). Together, these findings indicate that ablation of neurons that were overexpressing CREB at the time of memory-encoding blocks the memory of that particular learning event while leaving subsequent learning intact. Much like the data from wild-type mice infused with the mCREB vector (which showed normal memory), the high portion of remaining (noninfected) neurons seemed sufficient to encode a new memory. Finally, we examined the effects of ablating neurons that were infected with the CREB-cre vector after training. In this experiment, we reasoned that there would be no memory disruption because the CREB-infected neurons would not be part of the memory trace (because training had already taken place). This is exactly what we observed. Therefore, the memory loss induced by ablating neurons overexpressing CREB is robust, persistent and specific.
Our results show that the neurons with increased CREB levels at the time of fear learning are critical to the stability of that memory because selectively ablating just these neurons after training blocks this fear memory. This indicates that these neurons themselves are essential for later memory expression; they are not simply creating a local environment that promotes memory formation (such as releasing trophic factors). Fear learning may generate a broad memory trace that encompasses more lateral amygdala neurons than affected by our treatment or multiple memory traces throughout the brain. However, deleting just the neurons over-expressing CREB at the time of training produces amnesia, which suggests that these neurons play an essential role in what is likely a broader fear neuronal network.
Our results established a causal link between the activity of a defined subpopulation of neurons and expression of a fear memory, thereby identifying a key component of the memory trace. They also indicate that neurons with relatively high CREB levels are selectively recruited to a fear memory trace. But what is the mechanism underlying this preferential selection? Why are the neurons that overexpress CREB so special? One possibility is that increasing CREB alters the intrinsic excitability of a neuron. Intrinsic excitability (the propensity of a neuron to fire action potentials in response to an input) is determined by the distribution and properties of ion channels (e.g., Na+, K+, Ca2+) in the plasma membrane. Indeed, increasing CREB function stimulates transcription of a voltage-dependent Na+ subunit (1β subunit) and inhibits transcription of a voltage-dependent K+ channel subunit (Kv1.4).68 Hebb proposed that links between 2 cells are strengthened if both cells are active simultaneously.69 That is, coincident firing of the presynaptic neuron and depolarization of the postsynaptic neuron is necessary for Hebbian plasticity. A postsynaptic neuron that is more excitable than its neighbour might therefore be more likely to be depolarized and subsequently “fire together” and “wire together” with the presynaptic neuron. In this way, neurons with increased intrinsic excitability may be “primed” for learning and more likely to outcompete their neighbours for inclusion in a fear memory trace. Unlike synaptic plasticity, which involves changes at the level of the individual synapse, intrinsic excitability or plasticity involves changes at the level of the entire neuron,70 thus making it an attractive mechanism for the effects of CREB on neuronal competition during memory formation.
To investigate this, my laboratory collaborated with Mike Salter’s laboratory at The Hospital for Sick Children. We examined the electrophysiological characteristics of principal neurons in the lateral amygdala infected with CREBWT and Cntrl vectors in acute slices from wild-type mice. We were inspired by work from Eric Nestler’s and Robert Malenka’s laboratories71 showing that medium aspiny neurons in the nucleus accumbens infected with a similar CREB vector showed increased excitability (increased evoked action potential firing, decreased rheobase), whereas disrupting CREB (using the dominant-negative CREBS133A vector) decreased intrinsic excitability without affecting passive membrane properties.
We found a similar effect in lateral amygdala neurons. Our initial results showed that neurons with the CREBWT vector were more excitable than both their noninfected neighbours and those infected with the Cntrl vector.72 Specifically, neurons with the CREBWT vector fired more action potentials in response to the same input stimulus than neurons with the Cntrl vector or the noninfected neighbouring neurons. Importantly, we observed no change in passive membrane properties (average resting membrane potential, input conductance) or action potential waveforms between the groups. This increase in neuronal excitability is in agreement with findings using viral vectors73,74 and transgenic mice75–78 to increase CREB levels. Recently, the effects of increasing CREB function on the electrophysiological properties of lateral amygdala neurons has been replicated and extended by Alcino Silva’s group.79
Francis Crick wrote that a crucial step in understanding the mechanisms underlying memory is to interfere with defined neuronal populations in intact neural circuits.80 He emphasized that it was necessary that such manipulations target specific neurons in time and space. With the advent of modern genetic, molecular and imaging tools, this is becoming possible.81 Our studies have shown that interfering (retrograde deletion) with a defined neuronal population (lateral amygdala neurons overexpressing CREB) in intact circuits disrupts memory expression. This may be the first example of the disruption of a specific memory within a distributed network. We believe that this takes us one step closer to finding and even manipulating the elusive engram.
Acknowledgments
In this review, I outlined my scientific journey. Of course, I did not take this journey alone, as all the work described here has been a team effort. I mentioned many of the important people along the way. I would also like to specifically acknowledge certain individuals whose contributions were vital. First, my postdoctoral mentors, Mike Davis and Alcino Silva. Next, the members of my laboratories (both past and present), especially Jin-Hee Han, Adelaide Yiu, Christy Cole and Hwai-Lin (Liz) Hsiang. Finally, I thank my collaborators, Eric Nestler, Bill Carlezon, Rachael Neve and John Guzowski, and especially Steven Kushner and Paul Frankland. I would like to thank the people in my lab and my many collaborators for making this work possible. I would also like to thank Paul W. Frank-land for suggestions on this manuscript. This work was supported by Canadian Institutes of Health Research (MOP74650), EJLB Foundation and grants from the National Sciences and Engineering Research Council of Canada.
Footnotes
Competing interests: None declared.
- Received January 22, 2010.
- Revision received April 7, 2010.
- Accepted April 12, 2010.
References
In this issue

{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- Reactivating Hippocampal-Mediated Memories During Reconsolidation to Disrupt Fear
- CREB is Required in Excitatory Neurons in the Forebrain to Sustain Wakefulness
- Memory engrams: Recalling the past and imagining the future
- Example Based Hebbian Learning may be sufficient to support Human Intelligence
- Nonsynaptic plasticity model of long-term memory engrams
- Heroes of the Engram
- Emotional Modulation of Learning and Memory: Pharmacological Implications
- ZIP It: Neural Silencing Is an Additional Effect of the PKM-Zeta Inhibitor Zeta-Inhibitory Peptide
- Intrahippocampal Anisomycin Impairs Spatial Performance on the Morris Water Maze
- Engram cells retain memory under retrograde amnesia
- A Metaplasticity-Like Mechanism Supports the Selection of Fear Memories: Role of Protein Kinase A in the Amygdala
- Neurosilence: Profound Suppression of Neural Activity following Intracerebral Administration of the Protein Synthesis Inhibitor Anisomycin