

Abstract
Background: Major depressive disorder (MDD) is a global health concern. This study examined the efficacy, safety and tolerability of an extended-release (ER) formulation of levomilnacipran, an antidepressant approved for the treatment of MDD in adults.
Methods: This 10-week (1-week placebo run-in period, 8-week double-blind treatment, 1-week down-taper), multicentre, double-blind, placebo-controlled, parallel-group, fixed-dose study was conducted between June 2011 and March 2012. Adult outpatients (age 18–75 yr) with MDD were randomly assigned (1:1:1) to placebo or to levomilnacipran ER 40 mg/day or 80 mg/day. For primary efficacy, we analyzed the Montgomery–Åsberg Depression Rating Scale (MADRS) change from baseline to week 8 using a mixed-effects model for repeated-measures approach on the intent-to-treat (ITT) population. For secondary efficacy, we used the Sheehan Disability Scale (SDS), and for safety, we examined adverse events and laboratory, vital sign/physical and electrocardiography findings.
Results: The ITT population consisted of 185 patients in the placebo group, 185 in the levomilnacipran ER 40 mg/day group and 187 in the levomilnacipran ER 80 mg/day group. Study completion rates were similar among the groups (76%–83%). On MADRS change from baseline the least squares mean difference (LSMD) and 95% confidence interval (CI) versus placebo was significant for levomilnacipran ER 40 mg/day (–3.3 [−5.5 to −1.1], p = 0.003) and 80 mg/day (−3.1, [−5.3 to −1.0], p = 0.004). On SDS change from baseline the LSMD (and 95% CI) versus placebo was also significant for levomilnacipran ER 40 mg/day (−1.8, 95% [−3.6 to 0], p = 0.046) and 80 mg/day (−2.7 [−4.5 to −0.9], p = 0.003). More patients in the levomilnacipran ER than the placebo group prematurely exited the study owing to adverse events; common adverse events (≥ 5% and ≥ double the rate of placebo) were nausea, dry mouth, increased heart rate, constipation, dizziness, hyperhidrosis, urinary hesitation and erectile dysfunction.
Limitations: Limitations to our study included short treatment duration and lack of an active control arm.
Conclusion: Levomilnacipran ER at doses of 40 mg/day and 80 mg/day demonstrated efficacy on symptomatic and functional measures of MDD and was generally well tolerated in this patient population.
Clinical trial registration NCT01377194.
Introduction
Major depressive disorder (MDD) is a global health concern characterized by high lifetime prevalence and considerable disability.1 The disorder typically has a recurrent course, with at least half of patients who recover from a first episode of depression having 1 or more subsequent episodes.2 Patients with recurrent depression are likely to have more severe illness, anxiety, somatic and cognitive symptoms, and marked impairment.2,3 Given the substantial personal, societal and economic burdens associated with MDD, the development of new antidepressant agents is warranted to address patient needs that are not being met with current treatment options.
Levomilnacipran (1S, 2R-milnacipran) is a potent and selective serotonin and norepinephrine reuptake inhibitor (SNRI) that was approved in July 2013 for the treatment of MDD in adults; an extended-release (ER) formulation was developed to allow once-daily dosing. Levomilnacipran is the pharmacologically more active enantiomer of the racemic mixture milnacipran. In vitro studies have shown that levomilnacipran has approximately 2-fold greater potency for norepinephrine relative to serotonin reuptake inhibition.4 Relative to the less active enantiomer (F2696), levomilnacipran was 50 and 13 times more potent in inhibiting norepinephrine and serotonin reuptake, respectively; in the forced swim test, an animal model predictive of antidepressant efficacy, levomilnacipran was more than 30 times as potent as F2696. Levomilnacipran is distinct from the SNRIs duloxetine, venlafaxine and desvenlafaxine in that it preferentially inhibits norepinephrine reuptake; compared with these SNRIs, levomilnacipran is more than 10 times as selective for norepinephrine than serotonin reuptake inhibition.4,5
The efficacy of levomilnacipran ER for the treatment of MDD has been shown in 3 randomized, double-blind, placebo-controlled studies comparing fixed6 or flexible doses7,8 of levomilnacipran ER to placebo. In another flexible-dose study, levomilnacipran ER improved depressive symptoms, but did not achieve significant separation from placebo.9 Levomilnacipran ER was generally well tolerated in all 4 studies. We designed the present fixed-dose study to further examine the efficacy, safety and tolerability of levomilnacipran ER at doses of 40 mg/day and 80 mg/day in the treatment of adults with MDD.
Methods
This study (NCT01377194) was conducted at 51 centres in the United States and Canada between June 2011 and March 2012 in full compliance with U.S. Food and Drug Administration and Health Canada guidelines for Good Clinical Practice and in accordance with the ethical principles of the Declaration of Helsinki. The protocol was approved by the institutional review board at each study centre, and all patients provided written informed consent.
Study design
This 10-week multicentre, randomized, double-blind, placebo-controlled, parallel-group, fixed-dose study involved adult outpatients with MDD. The study consisted of a 1-week, single-blind placebo run-in period, an 8-week double-blind treatment period, and a 1-week, double-blind down-taper period. Patients who completed double-blind treatment or who prematurely exited the study were eligible to continue in the 1-week double-blind down-taper period if the investigators considered it medically appropriate; since human pharmacokinetic data have demonstrated a terminal half-life of 12 hours for levomilnacipran (data on file), we considered a down-taper period of 1 week to be sufficient to evaluate potential discontinuation symptoms.
Patients who met eligibility criteria were randomly assigned (1:1:1) to placebo, levomilnacipran ER 40 mg/day or levomilnacipran ER 80 mg/day. Allocation was performed using computer-generated randomization numbers, and treatment assignments were made using an interactive Web response system; randomization was not stratified by study centre. Identically appearing treatments with labels corresponding to the sequence of treatment assignment were supplied. Study personnel and patients were blinded to treatment group for the entire study period.
All patients assigned to the levomilnacipran ER groups received 20 mg on days 1–2 and 40 mg on days 3–5. Patients in the 40 mg/day group continued at this dose for the remainder of the study, whereas patients in the 80 mg/day group received a dose increase on day 6 and continued at 80 mg for the remainder of the study. All doses were taken once daily.
During the first 2 days of double-blind down-taper (week 9), the dose was maintained at 40 mg/day for patients in the 40 mg/day group and was decreased to 40 mg/day for patients in the 80 mg/day group; thereafter, the dose was decreased to 20 mg/day for all patients until the end of the down-taper period.
Inclusion criteria
Men and women aged 18–75 years who were outpatients and met the criteria for recurrent MDD as defined by the DSM-IV-TR10 were eligible to participate. Recurrent depression requires the presence of 2 or more major depressive episodes separated by an interval of at least 2 months, during which criteria for a major depressive episode are not met. We confirmed the diagnosis using the Mini International Neuropsychiatric Interview.11
Participants were also required to have an ongoing major depressive episode of 6 weeks to 12 months in duration, 5 or fewer major depressive episodes within the previous 5 years, score of 26 or higher on the clinician-rated Montgomery–Åsberg Depression Rating Scale (MADRS)12 and score of 4 or higher on the Clinical Global Impressions–Severity (CGI-S)13 scale. Physical criteria, including a negative serum β–human chorionic gonadotropin serum pregnancy test for female patients of childbearing potential, no clinically important findings on physical exam, clinical laboratory values or electrocardiogram (ECG) and body mass index of 18–40, were required.
Exclusion criteria
Patients with a DSM-IV-TR Axis I disorder other than MDD within 6 months of study or a lifetime history of other serious psychiatric disorders (e.g., manic episode/bipolar disorder, schizophrenia, severe personality disorder), or substance abuse or dependence within the 6 months preceding the study were excluded; secondary diagnoses of comorbid generalized and social anxiety disorders and/or specific phobias were allowed. Clinically relevant physical conditions (e.g., cardiovascular disease, history of cancer, concurrent medical condition that could interfere with the conduct of the study, pregnancy) and certain potentially confounding treatment-related criteria (e.g., initiation or termination of psychotherapy within the 3 months preceding the study, vagus nerve stimulation within the 6 months preceding the study) resulted in exclusion. Lifetime history of nonresponse to 2 or more antidepressants after adequate treatment trials or antidepressant nonresponse for the current depressive episode, use of prohibited concomitant medications (e.g., antidepressants, anticonvulsants, anti-psychotics, anxiolytics) and prior participation in an investigational study of milnacipran (indicated for the treatment of fibromyalgia in the United States) or levomilnacipran ER were also grounds for exclusion. Patients at high risk for suicide, defined as a suicide attempt within the past year, a score of 5 or higher on item 10 (Suicidal Thoughts) of the MADRS, a score of 3 or more on item 3 (Suicide) of the 17-item Hamilton Rating Scale for Depression (HAMD-17)14 or significant risk determined by the investigator interview or using the Columbia Suicide Severity Rating Scale (C-SSRS),15 could not participate.
Efficacy assessments
The primary efficacy measure was the MADRS, which was assessed at screening (week −1), baseline (week 0) and at weeks 1, 2, 4, 6 and 8. The secondary efficacy measure was the Sheehan Disability Scale (SDS),16 a measure of functional impairment assessing work, social life and family life (weeks 0, 4, 6 and 8). Additional end points included the HAMD-17 (weeks −1, 0, 4 and 8) and the CGI-S (weeks −1, 0, 1, 2, 4, 6 and 8).
Safety assessments
Adverse events, defined as any reported or observed untoward event that occurred from the time of informed consent until 30 days after the last treatment dose, were recorded (weeks 0, 1, 2, 4, 6, 8 and 9 [double-blind down-taper]) and evaluated in terms of intensity (mild, moderate, severe), seriousness and possible association with the study drug. Clinical laboratory tests (weeks −1, 4, 8 and 9), vital sign recordings (weeks −1, 0, 1, 2, 4, 6, 8 and 9) and 12-lead ECG examinations (weeks −1, 4 and 8) were conducted. The C-SSRS was administered at all study visits to assess the severity of suicidal behaviour and ideation; it was completed at screening to obtain lifetime history of suicidal ideation and behaviour and at weeks 0, 1, 2, 4, 6, 8 and 9 to evaluate ideation and behaviour since the previous visit.
Statistical analysis
The safety population was used for all safety analyses and consisted of all patients who were randomly assigned and received at least 1 dose of double-blind study drug. The intent-to-treat (ITT) population was used for all efficacy analyses and consisted of all patients in the safety population with least 1 MADRS total score assessment after baseline. We assessed demographic parameters using a 2-way analysis of variance. We compared reasons for discontinuation for each levomilnacipran ER dose group versus placebo using the Fisher exact test.
The primary efficacy parameter was change in MADRS total score from baseline to week 8. The primary analysis was conducted using a mixed-effects model for repeated-measures (MMRM) approach with treatment group, pooled study centre, visit and treatment group × visit interaction as fixed effects and the baseline score and baseline × visit interaction as the covariates. We used the Hochberg multiple-comparison procedure17 to control for type I errors. Sensitivity analyses were carried out using the last observation carried forward (LOCF) approach and the pattern-mixture model (PMM) approach based on non–future dependent missing value restrictions.18 The LOCF analysis was based on analysis of covariance with treatment group and pooled study centre as factors and the baseline MADRS total score as a covariate for between-group comparisons.
The secondary efficacy parameter was change in SDS total score from baseline to week 8, which we analyzed using an MMRM approach and a Hochberg multiple-comparison procedure similar to the primary efficacy measure. We also performed LOCF sensitivity analysis. The SDS total score was calculated only if scores for all 3 SDS domain subscales (Work, Family Life and Social Life) were available for analysis; the subscale domains were each calculated using non-missing observations.
Additional efficacy parameters included change from baseline to week 8 in SDS Work, Social Life and Family Life subscale scores, HAMD-17 total score and CGI-S score using MMRM and LOCF approaches similar to those used for the primary efficacy parameter. We analyzed the MADRS and HAMD-17 response (≥ 50% reduction in baseline total score) and remission (MADRS total score ≤ 10 and HAMD-17 total score ≤ 7) rates at week 8 using a logistic model with treatment group and corresponding baseline score as explanatory variables for the LOCF approach only.
Additional post hoc analyses evaluated SDS response (SDS total score ≤ 12 and all SDS subscale scores ≤ 4) and remission (SDS total score ≤ 6 and all SDS subscale scores ≤ 2)19,20 and MADRS total score change in patients stratified by median SDS score at baseline.
Laboratory, vital sign and ECG values were considered potentially clinically significant (PCS) if they met predefined high or low criteria. For safety parameters, no statistical analyses were done to compare treatment groups. All statistical tests were 2-sided hypothesis tests performed at the 5% level of significance for main effects; all confidence intervals (CIs) were 2-sided 95% CIs.
Results
Patient disposition and demographic characteristics
Patient disposition and reasons for premature exit from the study are presented in Table 1. Significantly more patients from the levomilnacipran ER 40 mg/day (p = 0.032) and 80 mg/day (p < 0.001) groups than the placebo group prematurely exited the study owing to adverse events. Treatment-emergent adverse events that resulted in discontinuation of 2 or more patients in 1 or both levomilnacipran ER groups were nausea, increased heart rate, rash and headache. No imbalances were observed among the treatment groups for any demographic or disease characteristic (Table 2); per protocol, all patients in the study had recurrent MDD.
Patient populations and disposition characteristics, by treatment group
Demographic and clinical characteristics of the safety population
Analysis of efficacy
Primary efficacy
Statistically significant and clinically meaningful improvement in change in MADRS total score from baseline to week 8 was demonstrated for both levomilnacipran ER 40 mg/day and 80 mg/day versus placebo using the primary MMRM analysis (least squares mean difference [LSMD] −3.3, p = 0.003 and −3.1, p = 0.004, respectively; Table 3). The difference in favour of levomilnacipran ER was statistically significant as early as 4 weeks after treatment initiation for both doses, and the significant difference persisted for the remainder of the 8-week double-blind treatment period (Fig. 1).
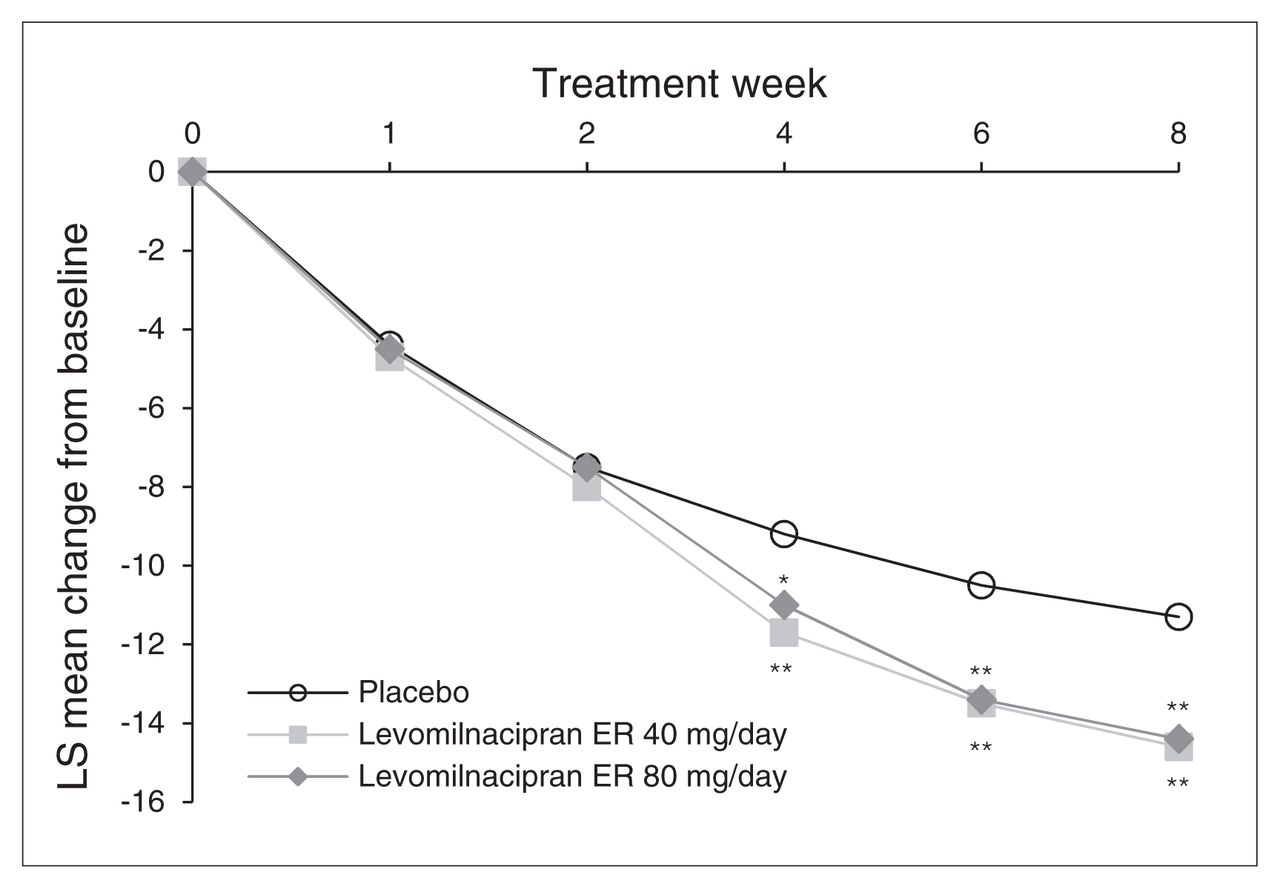
Change in Montgomery–Åsberg Depression Rating Scale (MADRS) total score from baseline to week 8 (intent-to-treat population, mixed-effects model for repeated-measures). *p < 0.05; **p < 0.01. ER = extended-release; LS = least squares.
Primary and secondary efficacy measures: change from baseline to week 8, intent-to-treat population
Results of the sensitivity analyses performed on MADRS change at week 8 confirmed the robustness of the primary MMRM results. Using the LOCF approach, a statistically significant difference versus placebo was observed for levomilnacipran ER 40 mg/day and 80 mg/day (Table 3). The PMM analyses demonstrated the robustness of the primary results to the possible violation of the missing-at-random assumption for both levomilnacipran ER treatment groups (Appendix, Table S1, available at cma.ca/jpn).
Secondary efficacy
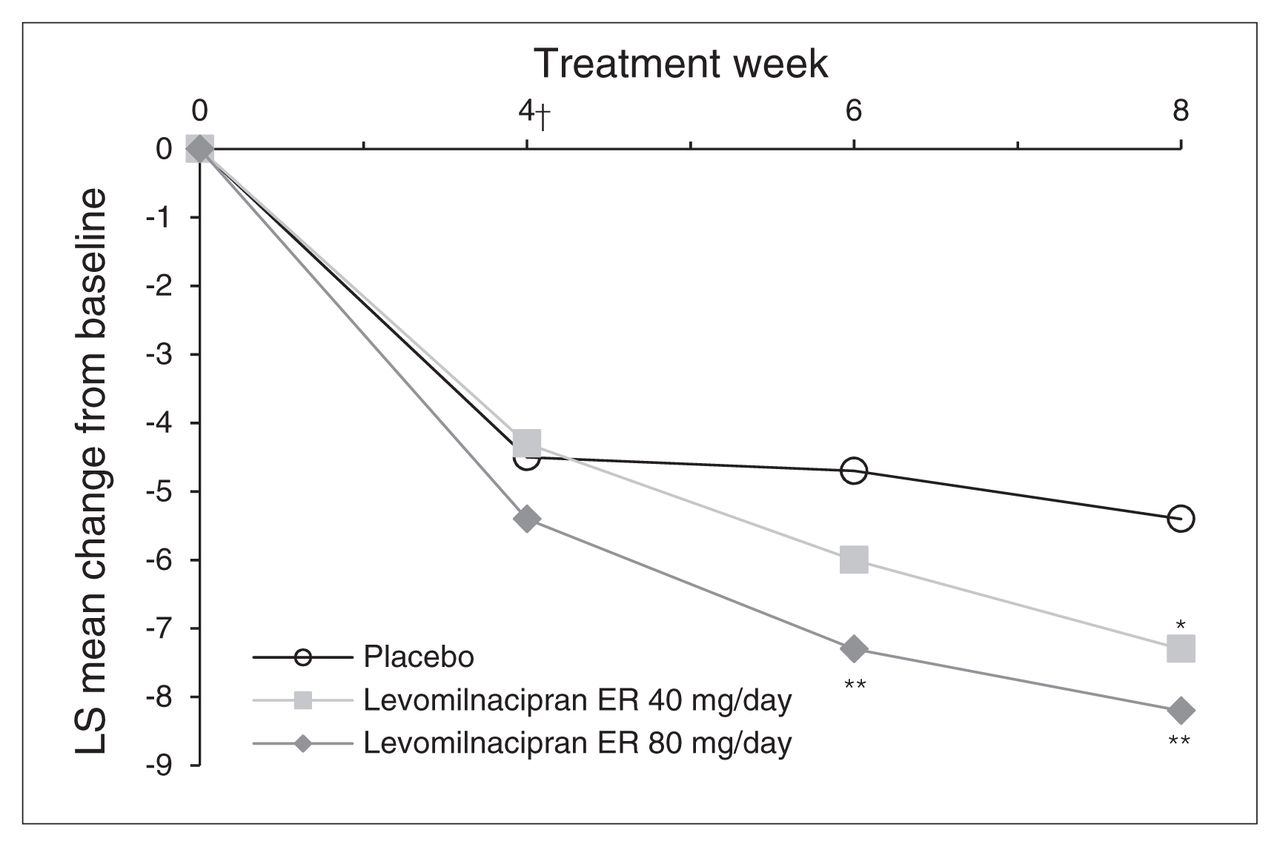
The SDS total score change from baseline to week 8 was significantly greater for levomilnacipran ER 40 mg/day and 80 mg/day treatment groups than the placebo group (MMRM); using the LOCF approach, the difference in SDS total score was statistically significant for the 80 mg/day dose (Table 3). The SDS total scores improved at every study assessment for both doses of levomilnacipran ER; statistically significant separation from placebo occurred at week 6 for the levomilnacipran ER 80 mg/day group (Fig. 2). Change from baseline on the SDS Work subscale was significantly greater for both dose groups relative to the placebo group; no significant differences versus placebo were observed on the Social Life and Family Life subscales (Table 3).

Change in Sheehan Disability Scale (SDS) total score from baseline to week 8 (intent-to-treat population, mixed-effects model for repeated-measures). *p < 0.05; **p < 0.01. †First postbaseline assessment of the SDS. ER = extended-release; LS = least squares.
Additional efficacy
Improvement in HAMD-17 total score from baseline to week 8 (MMRM) was significantly greater for the levomilnacipran ER 40 mg/day and 80 mg/day groups than the placebo group; the LSMD versus placebo was −2.2 (95% CI −3.7 to −0.6, p = 0.007) for the 40 mg group and −1.6 (95% CI −3.2 to −0.1, p = 0.043) for the 80 mg group. Change from baseline to week 8 (MMRM) in CGI-S scores was also significantly greater for both levomilnacipran ER dose groups than placebo, as shown by the LSMD: −0.3 (95% CI −0.6 to 0.0, p = 0.020) in the 40 mg group and −0.3 (95% CI −0.6 to −0.1, p = 0.015) in the 80 mg group.
In a post hoc analysis of the subset of patients with greater functional impairment (defined as baseline SDS score greater than the median baseline score of 17), MADRS total score change was significantly greater for levomilnacipran ER than placebo (MMRM) in the 80 mg group (LSMD −4.5, 95% CI −8.4 to −0.7, p = 0.022), but not in the 40-mg group (LSMD −1.3, 95% CI −5.4 to 2.6, p = 0.50).
A greater percentage of patients in the levomilnacipran ER 40 mg/day (49%, p = 0.004) and 80 mg/day (47%, p = 0.010) groups than in the placebo group (34%) achieved MADRS response (≥ 50% improvement); rates of MADRS remission (MADRS ≤ 10) were also significantly greater for levomilnacipran ER 40 mg/day (30%, p = 0.012) and 80 mg/day (32%, p = 0.002) than placebo (18%; Fig. 3). A HAMD-17 response (≥ 50% improvement) was achieved by a significantly greater percentage of patients in the levomilnacipran ER 40 mg/day group (45%, p = 0.009) than in the placebo group (31%); no significant difference between levomilnacipran ER 80 mg/day (39%) and placebo (p = 0.10) was noted. Significantly greater HAMD-17 remission (score ≤ 7) was observed in the levomilnacipran ER 40 mg/day (30%, p = 0.028) and 80 mg/day (30%, p = 0.023) groups than in the placebo group (20%).

Montgomery–Åsberg Depression Rating Scale (MADRS) response and remission (intent-to-treat population, last observation carried forward). *p < 0.05; **p < 0.01. ER = extended-release.
We also noted significant improvements for levomilnacipran ER relative to placebo in response and remission using functional outcome measures. The SDS response rates (SDS total score ≤ 12 and all SDS subscale scores ≤ 4) were significantly greater for levomilnacipran ER 40 mg/day (57%, p = 0.008) and 80 mg/day (61%, p < 0.001) than placebo (42%). Similarly, SDS remission rates (SDS total score ≤ 6 and all SDS subscale scores ≤ 2) were significantly higher for levomilnacipran ER 40 mg/day (32%, p = 0.048) and 80 mg/day (32%, p = 0.031) than placebo (21%; Fig. 4).
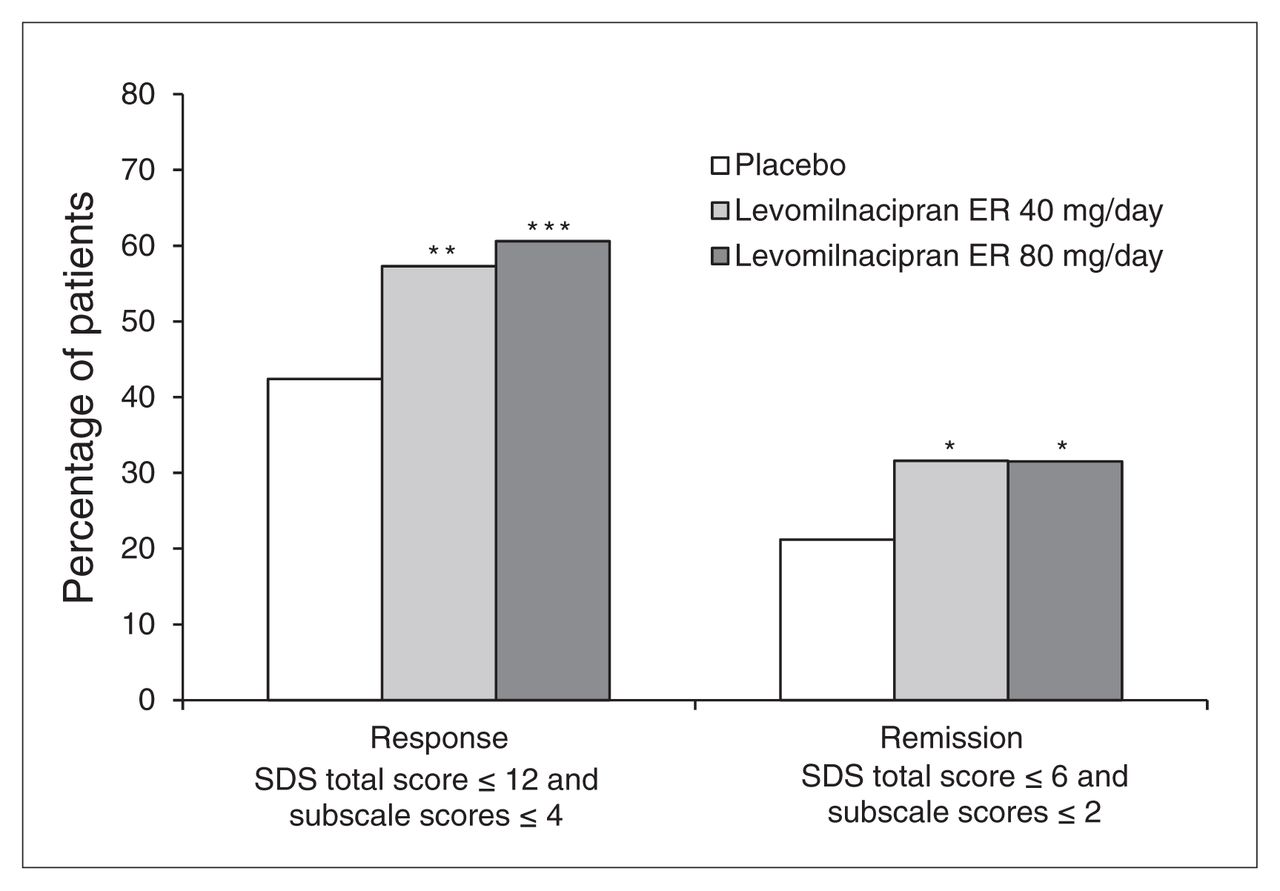
Sheehan Disability Scale (SDS) response and remission (intent-to-treat population, last observation carried forward). *p < 0.05; **p < 0.01; ***p < 0.001. ER = extended-release.
Analysis of safety
Adverse events
An overall summary of adverse events is presented in Table 4. During double-blind treatment, serious adverse events were reported for 1 patient in the placebo group (facial bones fracture/road traffic accident) and 3 patients in the levomilnacipran ER 40 mg/day group (noncardiac chest pain, n = 1; intussusception, n = 1; asthma, n = 1). Two patients in the levomilnacipran ER 40 mg/day group exited the study owing to serious adverse events (intussusception, n = 1, and asthma, n = 1). No serious adverse events were reported in the levomilnacipran ER 80 mg/day group, and no serious adverse event was considered by the investigator to be treatment-related.
Summary of adverse events, safety population
The treatment-emergent adverse events that were reported in 5% or more patients in any treatment group during double-blind treatment are presented in Table 5. Investigators considered most treatment-emergent adverse events to be mild or moderate in intensity, and in all 3 treatment groups most were considered to be related to treatment. Treatment-emergent adverse events that occurred at an incidence of 5% or more and at least at twice the rate of placebo in both levomilnacipran ER groups were nausea, dry mouth, constipation, increased heart rate, dizziness, hyperhidrosis, urinary hesitation and erectile dysfunction; in the 80 mg/day group, increased heart rate and testicular pain also occurred at this rate. The incidence of newly emergent adverse events during the 1-week double-blind down-taper was low in all treatment groups. During the down-taper period, no treatment-emergent adverse events were considered to be severe, and most events in all groups were not considered to be related to treatment.
Common treatment-emergent adverse events (≥ 5% of patients in any treatment group), safety population
Laboratory values
For placebo, levomilnacipran ER 40 mg and levomilnacipran ER 80 mg, the mean ± standard deviation change from baseline in alanine aminotransferase (ALT) was 0.0165 ± 0.1579 μkat/L (1.0 ± 9.5 U/L), 0.0295 ± 0.1398 μkat/L (1.8 ± 8.4 U/L) and 0.0765 ± 0.3430 μkat/L (4.6 ± 20.5 U/L), respectively; mean change in aspartate aminotransferase (AST) was 0.0077 ± 0.1099 μkat/L (0.5 ± 6.6 U/L), 0.0280 ± 0.0859 μkat/L (1.7 ± 5.1 U/L) and 0.0499 ± 0.2515 μkat/L (3.0 ± 15.1 U/L), respectively; and mean change in alkaline phosphatase was −0.0289 ± 0.1530 μkat/L (−1.7 ± 9.2 U/L), 0.0486 ± 0.1280 μkat/L (2.9 ± 7.7 U/L) and 0.0370 ± 0.1607 μkat/L (2.2 ± 9.6 U/L), respectively. No patients in the levomilnacipran ER 40 mg group met PCS criteria for ALT or AST (≥ 3 × upper limit of normal [ULN]); in the levomilnacipran ER 80 mg group, 2 (1.1%) patients met PCS criteria for ALT and 2 (1.1%) patients met PCS criteria for AST. Two patients had PCS laboratory values that were reported as treatment-emergent adverse events (bilirubin increase in 1 patient in the 40 mg group, and ALT and AST in 1 patient in the 80 mg group); no patients met the criteria for Hy’s Law (ALT or AST ≥ 3 × ULN and total bilirubin > 2 × ULN in the absence of alkaline phosphatase > 2 × ULN).21
Vital signs and orthostatic hypotension
Blood pressure and pulse rate increases were similar for both levomilnacipran ER doses, but they were higher relative to placebo (Table 6). The overall incidence of orthostatic hypotension was generally similar among the groups and was not indicative of meaningful change; no patients exited the study as a result of orthostatic hypotension. The PCS changes in vital signs and weight were relatively low and similar among the groups (Table 6).
Mean changes and incidence of potentially significant vital sign parameters, safety population
Electrocardiography
Mean changes in ECG parameters are presented in Table 7. Mean increases in QTcB interval in the levomilnacipran ER treatment groups were consistent with increases in ventricular heart rate; mean QTcF interval changes did not demonstrate any signal. No patients in any treatment group had a treatment-emergent QTcB or QTcF value greater than 500 ms.
Mean changes in electrocardiographic parameters at the end of the double-blind treatment, safety population
Measures of suicidality
Suicidal ideation and behaviour
During double-blind treatment, the incidence of C-SSRS–rated suicidal ideation was similar for placebo (11.4%) and levomilnacipran ER 80 mg/day (14.4%), and somewhat greater for levomilnacipran ER 40 mg/day (19.5%). Most events were classified as the least severe suicidal ideation (“wish to be dead,” without intent to act) for placebo (7.6%), levomilnacipran ER 40 mg/day (15.7%) and levomilnacipran ER 80 mg/day (10.1%). No patients demonstrated suicidal behaviour during double-blind treatment or down-taper. The incidence of suicidal ideation was low and similar in all treatment groups during the down-taper period (6% in the placebo group and 4% in both levomilnacipran ER treatment groups).
Suicide-related treatment-emergent adverse events
During double-blind treatment, suicidal ideation was reported as a treatment-emergent adverse event in 1 patient in the placebo group and 1 patient in the levomilnacipran ER 80 mg/day group. Neither event was considered a serious adverse event; no patients exited the study owing to suicide-related treatment-emergent adverse events. No instances of suicide attempt or other suicidal behaviour were reported.
Discussion
We observed a significant reduction in MADRS total score starting at week 4 in patients treated with levomilnacipran ER 40 mg/day or 80 mg/day compared with placebo. For both doses of levomilnacipran ER, the treatment advantage versus placebo exceeded 2 points on the MADRS, suggesting that symptom improvement was clinically relevant.22
Both doses of levomilnacipran ER compared with placebo also significantly reduced SDS total score from baseline, indicating that functional improvement accompanied symptomatic improvement in our study. Synchronous symptomatic and functional improvement is noteworthy, because functional improvement often lags behind symptomatic improvement and patients with marked symptomatic improvement, especially those with chronic and severe depression, may continue to experience functional impairment.23 Because greater functional impairment negatively impacts personal and economic well-being and lowers the likelihood of recovery,24 it is recommended that patient wellness should be determined using both symptomatic and functional measures of remission.23
In a previous fixed-dose study of levomilnacipran ER 40 mg/day, 80 mg/day and 120 mg/day,6 numerical dose-related improvements were reported for most efficacy outcomes. We observed significant differences in favour of levomilnacipran ER 40 mg/day and 80 mg/day versus placebo on most additional efficacy parameters in the present study, although the magnitude of improvement for levomilnacipran ER 40 mg and 80 mg was generally similar. Of note, we observed numerically greater improvement in patients with higher baseline functional impairment (baseline SDS > 17).
Rates of MADRS response and remission were significantly higher for patients taking levomilnacipran ER than those taking placebo, with the difference for both doses versus placebo exceeding the 10% threshold typically regarded as clinically relevant in studies submitted for regulatory approval.22,25 The rate of SDS remission (total score ≤ 6 and all subscale scores ≤ 2) was also statistically significant for both levomilnacipran ER dose groups versus placebo, indicating that functional improvement occurred in conjunction with symptomatically relevant clinical change and suggesting synchronous functional and symptomatic improvement. In addition, because recurrent depressive episodes have been associated with failure to reach sufficient remission after adequate treatment,26 remission rates in the present study are of particular relevance because all patients had recurrent depression and remission was reached within an acute 8-week time frame.
Good safety and tolerability are factors in patients remaining on treatment long enough to fully benefit from it. Since many patients do not achieve lasting remission from MDD with short-term treatment, receiving treatment of adequate dose and duration is important.23 In the present study, levomilnacipran ER was generally well tolerated, with high study completion rates across groups. More patients in the levomilnacipran ER groups than the placebo group prematurely exited the study owing to adverse events; however, most common treatment-emergent adverse events were considered to be mild or moderate in intensity and consistent with adverse events commonly associated with SNRI treatment and noradrenergic effect.27 The incidence of serious adverse events was low, and no serious adverse events were considered to be related to the study drug.
Laboratory assessments showed slightly higher mean values for the levomilnacipran ER groups than the placebo group for some analytes, but there were no clinically meaningful differences among the groups. Increased pulse rate and blood pressure, most likely due to increased noradrenergic effects, occurred with levomilnacipran ER treatment; PCS incidence of increased pulse rate and blood pressure were low and similar among groups. We observed increases in QTcB interval in the levomilnacipran ER treatment groups; however, QTcB interval is known to overcorrect at higher heart rates, making measurement of QT prolongation with this correction problematic when assessing drugs that increase heart rate.28,29 Mean QTcF interval increases in the levomilnacipran ER groups were low and similar to that in the placebo group. No patient had PCS increases (> 500 ms) in either QTcB or QTcF interval.
Limitations
Full interpretation of our study results is limited by the short treatment duration and the lack of an active control arm. Because SDS total score can be computed only when all 3 subscales are available, the measurable difference in SDS functioning applies only to patients with complete SDS assessments. In this study, about 30% of patients did not have a Work/School subscale score, so SDS results apply only to the 70% of patients who were working or going to school at baseline. Our inclusion and exclusion criteria may have limited the ability to generalize the findings to a wider population. To participate, patients were required to have recurrent MDD, a very common disease course, but one that may not be relevant to all patients with MDD. Unlike some other trials, our trial excluded patients with nonresponse to adequate treatment with an antidepressant during the current depressive episode.
Conclusion
This 8-week, fixed-dose, phase III study of levomilnacipran ER 40 mg/day or 80 mg/day compared with placebo demonstrated significant improvement in symptoms of depression and functional impairment associated with MDD. On the primary efficacy measure, statistically significant differences compared with placebo that persisted for the remainder of the study were apparent 4 weeks after treatment initiation in both dose groups. Treatment with levomilnacipran ER was generally well tolerated, and the adverse event profile was similar to those reported in prior levomilnacipran ER studies. Our results, in conjunction with the robust positive outcomes in 3 of 4 previous clinical trials, support the use of levomilnacipran ER as an effective treatment for symptomatic and functional impairment associated with MDD.
Acknowledgements
This study was supported by funding from Forest Laboratories, Inc. (New York, New York). Forest Laboratories, Inc. was involved in the study design, collection (via contracted clinical investigator sites), analysis and interpretation of data, and the decision to present these results. Writing assistance and editorial support for the preparation of this manuscript was provided by Carol Dyer, MS, and Adam Ruth, PhD, of Prescott Medical Communications Group, Chicago, Illinois, contractors of Forest Research Institute.
Footnotes
Competing interests: D. Bakish is a recipient of grants as a principal investigator from Forest Laboratories and serves on the speaker’s bureau for Pfizer. M.L. Liebowitz has received grants from Pfizer, an investigator research contract from Forest Laboratories, and has stock options in Pherin Pharmaceuticals. A. Khan is the founder and Medical Director of Columbia Northwest Pharmaceuticals LLC and owns intellectual property rights for potential therapies for central nervous system disorders and other medical conditions. C.P. Gommoll, C. Chen, R. Nunez, and W.M. Greenberg are employees of Forest Research Institute, a subsidiary of Forest Laboratories. A. Bose was an employee of Forest Research Institute at the time of study and is currently employed by Otsuka Pharmaceutical Development & Commercialization.
Contributors: A. Bose, C. Gommoll, R. Nunez and W.M. Greenberg designed the study. D. Bakish acquired the data, which all authors analyzed. A. Bose and W.M. Greenberg wrote the article, which all authors reviewed and approved for publication.
- Received March 6, 2013.
- Revision received April 22, 2013.
- Revision received June 27, 2013.
- Accepted July 2, 2013.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.