

Abstract
Background: Although previous study has demonstrated that brain-derived neurotrophic factor (BDNF) is involved in the antidepressant-like effect of oleanolic acid, there is little information regarding the details of the molecular mechanism involved in this effect.
Methods: We used a chronic unpredictable mild stress (CUMS) model to test the antidepressant-like effect of oleanolic acid on depressant-like behaviour, miR-132 expression and synaptic protein expression in the male mouse hippocampus. Furthermore, we explored the possible signalling pathways associated with miR-132 expression that mediate the effect of oleanolic acid on neuronal proliferation.
Results: The results demonstrated that a 3-week treatment with oleanolic acid ameliorated CUMS-induced anhedonic and anxiogenic behaviours. Furthermore, we found that oleanolic acid led to the BDNF-related phosphorylation and activation of extracellular signal-regulated kinases (ERK) and cyclic adenosine monophosphate response element binding protein (CREB), which was associated with the upregulation of miR-132 and hippocampal neuronal proliferation. Moreover, experiments with an miR-132 antagomir revealed that targeting miR-132 led to inhibition of neuronal proliferation and the postsynaptic density protein 95, but did not affect presynaptic protein synapsin I.
Limitations: Several other stimuli can also induce CREB phosphorylation in the hippocampus. Thus, regulation of miR-132 may not be restricted to neurotrophic signalling.
Conclusion: Our results show that oleanolic acid induces the upregulation of miR-132, which serves as an important regulator of neurotrophic actions, mainly through the activation of the hippocampal BDNF–ERK–CREB signalling pathways.
Introduction
Depression has become an increasingly prevalent health problem throughout the world. It is generally assumed that multiple mechanisms are responsible for the development of depression. Although clinical and experimental studies are providing some insight into the pathophysiological processes that may occur in depression, recent analyses have indicated that approximately 30% of depressive patients failed to respond satisfactorily to commercially available antidepressants in monotherapy.1 A growing body of evidence supports that brain-derived neurotrophic factor (BDNF) and its tropomyosin-related kinase receptor B (TrkB) are involved in the pathophysiology and treatment of depression.2,3 They play a critical role in the modulation of some functions, such as neurotransmitter release and postsynaptic responses to neurotransmitters, which are closely related to antidepressant therapy.4 Either reduced BDNF availability or decreased expression of the TrkB receptor could reduce BDNF–TrkB signalling.5 In contrast, treatment with antidepressants could upregulate BDNF or activate the TrkB receptor in the brain.6,7 In addition, in an animal model of depression, the results demonstrate that antidepressant efficacy is mediated at least in part through an elevation of BDNF levels or BDNF–TrkB signalling in the hippocampus.8,9 In contrast, in transgenic animals with decreased brain BDNF levels or inhibited BDNF–TrkB signalling, antidepressant agents fail to exert behavioural responses.10,11 Collectively, these studies indicate that stimulation of the BDNF–TrkB signalling pathway could provide a novel approach to the treatment of depression.
MicroRNAs (miRNAs) are small noncoding RNA molecules (about 22 nucleotides) found in plants and animals that function in the regulation of gene expression. Dysfunctional microRNA-mediated regulation has been implicated in the pathogenesis of many disorders, including diseases of the central nervous system. Neural miRNAs are involved at various stages of synaptic development, including dendritogenesis, synapse formation and synapse maturation. In mammals, miR-132 is enriched in neurons. The role of miR-132 in neuronal outgrowth and synaptic function is currently being studied. It has been suggested that the expression of miR-132 is induced by neurotrophins, such as BDNF,12,13 and this may represent a mechanism of fine-tuning the protein expression following neurotrophic stimulation.14 Specifically, by decreasing the levels of p250GAP, a guanosine triphosphatase activating protein linked to neuronal differentiation, miR-132 promotes neuronal outgrowth and brings about an increase in synaptic protein levels.15,16
Oleanolic acid, a pentacyclic triterpene, has been reported to induce serotonin (5-HT) secretion17 and alleviate cerebral ischemic damage.18 Our previous study also demonstrated that BDNF was involved in the antidepressant-like effect of oleanolic acid.19 However, there is little information regarding the details of the molecular mechanism involved in these effects. We therefore investigated whether the administration of oleanolic acid would lead to enhanced BDNF-related miR-132 expression in a mouse model of chronic unpredictable mild stress (CUMS) and thereby explored the major intracellular pathways activated by oleanolic acid treatment.
Methods
Animals
Male ICR mice (24 ± 2 g; 5 weeks old) were purchased from Shanghai Slac Animal Center, China. Animals were housed 8 per cage (320 × 180 × 160 cm) under a normal 12-hour light/dark schedule with the lights on at 7 am. The animals were allowed 1 week to acclimatize to the housing conditions before the beginning of the experiments. Ambient temperature and relative humidity were maintained at 22 ± 2°C and at 55 ± 5%, respectively, and the animals were given free access to standard chow and water for the duration of the study. All procedures were performed in accordance with the published guidelines of the China Council on Animal Care (Regulations for the Administration of Affairs Concerning Experimental Animals, approved by the State Council on Oct. 31, 1988, and promulgated by Decree No. 2 of the State Science and Technology Commission on Nov. 14, 1988). Each experimental group consisted of 8 mice, and the present study involved a total of 208 mice. All procedures were approved by the Institute for Experimental Animals of Huaqiao University.
Chemicals
We obtained oleanolic acid (purity = 98.4% by high- performance liquid chromatography) from Shanxi Huike Botanical Development Co., Ltd. Fluoxetine hydrochloride and bromodeoxyuridine (BrdU) were purchased from Sigma, and K252a was purchased from Alomone Laboratories. The miR-132, U6 primers and related miRNA reagents were purchased from RiboBio Co., Ltd. The anti-BDNF antibody was purchased from Santa Cruz Biotechnology Inc. The antibodies for BrdU and synapsin I were purchased from Millipore. The inhibitor U0126, and the antibodies for extracellular signal-regulated kinases (ERK)-1/2, phospho-ERK1/2, cyclic adenosine monophosphate response element binding protein (CREB), phospho-CREB and postsynaptic density protein 95 (PSD95) were purchased from Cell Signaling Technology. The anti–glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was purchased from Kangcheng Biotech.
Drug administration
For the test of the behavioural and miR-132 regulations, the mice were randomly divided into 14 groups: 7 control groups and 7 CUMS groups that received vehicle (0.9% physiologic saline); 20 mg/kg of fluoxetine (20 mg/kg); or 2.5, 5, 10, 20 or 40 mg/kg of oleanolic acid, respectively. Oleanolic acid was suspended in saline with 10% (v/v) Tween-80, and fluoxetine was dissolved in 0.9% physiologic saline. Oleanolic acid and fluoxetine were administered by oral gavage. The doses chosen and the administration method were based on the behavioural results and previous reports.19
To observe whether TrkB receptor activation is required for miR-132, we randomly divided the mice into 4 groups: the control-vehicle group, the CUMS-vehicle group, the CUMS-oleanolic acid group (20 mg/kg) and the CUMS-oleanolic acid+K252a group (20 mg/kg+25 μg/kg). We dissolved K252a in 0.1% dimethyl sulfoxide (DMSO) and saline and injected it intraperitoneally in a volume of 10 mL/kg body weight. The dose of K252a was selected based on previous studies.20–22
To ascertain whether ERK activation is a major contributor to miR-132 regulation by oleanolic acid, we randomly divided the mice into 4 groups: the control-vehicle group, the CUMS-vehicle group, the CUMS-oleanolic acid group (20 mg/kg) and the CUMS-oleanolic acid+U0126 group (20 mg/kg + 0.4 mg/kg). We dissolved U0126 in 0.1% DMSO and saline and injected it intravenously in a volume of 10 mL/kg body weight.23,24
The drug treatment was performed once daily for the last 3 weeks of the experiments.
Chronic unpredictable mild stress
We performed the CUMS procedure as described previously.25 Briefly, the weekly stress regime consisted of food and water deprivation, exposure to an empty bottle, exposure to a soiled cage, light/dark succession every 2 hours, space reduction, 45° cage tilt, overnight illumination and predator sounds. All stressors were applied individually and continuously, day and night. The control animals were housed in a separate room and had no contact with the stressed animals. To prevent habituation and ensure the unpredictability of the stressors, all the stressors were randomly scheduled over a 1-week period and repeated throughout the 7-week experiment. On the basis of their sucrose preference following 4 weeks of CUMS, both stressed and control mice were divided into matched subgroups.
Sucrose preference test (SPT)
The SPT was carried out after 4 weeks and 7 weeks of CUMS exposure. Briefly, before the test, the mice were trained to adapt to sucrose solution (1%, w/v): 2 bottles of sucrose solution were placed in each cage for 24 hours, and then 1 bottle of sucrose solution was replaced with water for 24 hours. After the adaptation, the mice were deprived of water and food for 24 hours. The test was conducted at 9:30 am, and during the test the mice were housed in individual cages and had free access to 2 bottles containing sucrose solution and water, respectively. After 24 hours, we recorded the volumes of the sucrose solution and water that had been consumed.
Novelty-suppressed feeding test (NSFT)
The NSFT was conducted 24 hours after the last SPT. The NSFT was performed during an 8-minute period, as previously described.26,27 Briefly, the testing apparatus consisted of a plastic box (50 × 50 × 20 cm). Food was withheld from the mice for 24 hours before the test. At the beginning of the test, a single pellet of food was placed on a white paper platform positioned at the centre of the box. We placed a mouse in a corner of the maze and immediately started a stopwatch. The scoring to measure interest began when the mouse reached for the food with its forepaws and began eating. As a control value, we measured the home-cage food consumption within the 15 minutes immediately following the test.
Real-time polymerase chain reaction (PCR)
To detect the relative protein or miR-132 expression, 5 animals per group were anesthetized and sacrificed 24 hours after the NSFT. The whole hippocampus was carefully dissected from each hemisphere, frozen in liquid nitrogen and stored at –80°C. We used 1 hemisphere for the PCR experiment and the other for a Western blot experiment. Four samples were randomly selected from 5 hippocampus tissues and used for each experiment.
We extrated total RNA using the miRNeasy kit (QIAGEN). We used Bulge-Loop miRNA primers for the mouse miR-132 (RiboBio), and real-time PCR reactions were performed using a SYBR Premix Ex Taq Kit in the Applied Biosystems 7500 system. We analyzed the results using the 2–ΔΔCT method. The expression of the U6 small nucleolar RNA gene was used as an internal control.
miR-132 antagomir experiments
To study the potential function of miR-132 in mouse hippocampal neuronal proliferation, we adopted a miRNA antagomir strategy. We antagonized the expression of miR-132 in the hippocampus using an antagomir that specifically targets miR-132. Mice were randomly divided into 4 groups: the control-vehicle group, the CUMS-vehicle group, the CUMS-oleanolic acid+antagomir-control group and the CUMS- oleanolic acid+miR-132 antagomir group. A miR-132 antagomir or an antagomir-control was dissolved in artificial cerebrospinal fluid (0.5 nmol for each mouse) and infused by micro-syringe into the lateral ventricle.28,29 The repeated antagomir treatment was performed once per week during the last 3 weeks of the experiment.
Western blot
Hippocampus samples were homogenized in a lysis buffer and incubated on ice for 30 minutes. The homogenates were centrifuged at 14 000g for 20 minutes at 4°C, and the supernatants were collected. The protein concentration was determined by a bicinchoninic acid assay. The proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. After blocking in 3% bovine serum albumin/tris-buffered saline with Tween 20 (TBST) at room temperature for 1 hour, the membranes were incubated with the appropriate primary antibodies at 4°C overnight (anti-BDNF 1:500, anti-synapsin I 1:1000, anti-PSD95 1:1000, anti-ERK 1:1000, anti-pERK 1:2000, anti-CREB 1:1000, anti-pCREB 1:1000, anti-GAPDH 1:5000). After washing with TBST 3 times, the membranes were incubated with an horseradish peroxidase–labelled secondary antibody (1:2000). The blots were washed again 3 times with TBST buffer, and the immunoreactive bands were detected using the enhanced chemiluminescence method. The results were normalized to the protein expression level of GAPDH in each sample.
Brain extraction and immunohistochemistry
For the test of the hippocampal neuronal proliferation, 3 mice were injected with BrdU (50 mg/kg, twice daily for 2 successive days). Twenty-four hours after the last BrdU injection, the mice were immediately anesthetized with chloral hydrate (0.35 g/kg) and then sacrificed by intracardial perfusion with heparinized 0.9% saline followed by ice-cold 4% paraformaldehyde. The brains were removed and postfixed with 4% paraformaldehyde overnight and incubated with 30% sucrose solution in phosphate-buffered saline (PBS) at 4°C for 2 days.
To determine the number of BrdU-incorporating cells in the hippocampus, 6 coronal sections (40 μm) were randomly selected throughout the hippocampus 1.70–2.57 mm posterior to the bregma, as defined in the mouse brain atlas, and the sections were analyzed by immunohistochemistry.30 Free-floating brain sections were incubated for 15 minutes in 0.3% hydrogen peroxide at room temperature to eliminate endogenous peroxidase. To detect BrdU-incorporated cells, the free-floating sections were rinsed extensively with PBS (0.05 M), and the sections were incubated for 30 minutes in hydrogen chloride at 37 °C to denature the DNA. The sections were neutralized by incubating them for 10 minutes in borate buffer. Then we used the anti-BrdU antibody as a primary antibody (1:5000) overnight at room temperature. Subsequently, the sections were intermittently rinsed with PBS and incubated with a biotinylated anti-rat immunoglobulin G secondary antibody (1:200) followed by avidin-biotin complex (1:100) for 1 hour at room temperature. BrdU-incorporated cells were visualized with 0.02% 3,3′-diaminobenzidine tetrahydrochloride and 0.01% hydrogen peroxide in 0.05 M PBS for approximately 3 minutes. The total number of BrdU-labelled cells per section was determined and added to obtain the total number of cells per animal.
Statistical analysis
All data are expressed as means ± standard errors of the mean. We analyzed the data using 1-way analysis of variance, followed by a 2-tailed Dunnett post hoc test. We considered results to be significant at p < 0.05.
Results
Oleanolic acid restores the behavioural dysfunctions induced by CUMS
After a 4-week CUMS procedure, the sucrose preference in the CUMS groups was significantly lower than that in the control groups for the first SPT (77.03 ± 2.44 in the control groups v. 52.71 ± 2.40 in the CUMS groups, F1,110 = 50.214, p < 0.001). Treatment with oleanolic acid for 3 weeks at concentrations of 5–40 mg/kg significantly increased the sucrose preference in the SPT and decreased the latency period until feeding in the NSFT; the change at a concentration of 2.5 mg/kg was not significant (Fig. 1A and B). The largest effect was obtained at a concentration of 20 mg/kg, when the sucrose preference increased by 72.8% (81.76 v. 47.32) and the latency period until feeding decreased by 40.4% (223.25 v. 386.87) relative to the CUMS-vehicle group. On the other hand, there was no difference in the home-cage feed consumption within 15 minutes (Fig. 1C) conducted immediately after the NSFT, indicating that the effects of oleanolic acid were not due to a general increase in feeding.
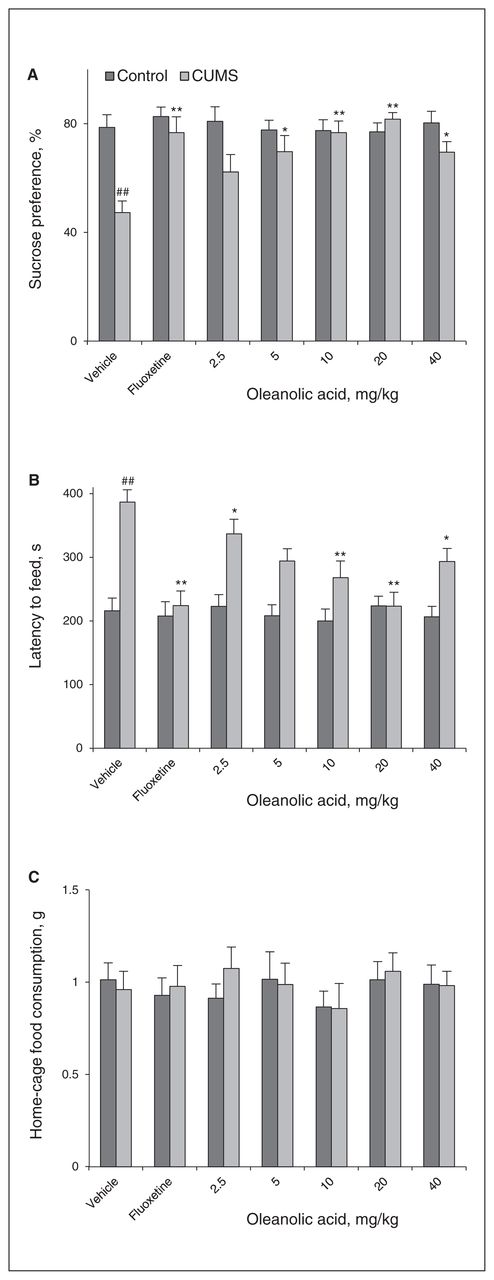
Oleanolic acid produces antidepressant-like effect in the chronic unpredictable mild stress (CUMS) model. The behavioural tests were conducted 1 hour after the last injection. (A) Oleanolic acid increased the sucrose preference in the CUMS groups (F6,49 = 5.698, p < 0.001). (B) Oleanolic acid decreased the latency period until feeding in the novelty-suppressed feeding test (NSFT; F6,49 = 7.322, p < 0.001). (C) Oleanolic acid had no effect on the home-cage feed consumption within 15 minutes following the NSFT (F6,49 = 0.44, p = 0.85) Oleanolic acid did not affect sucrose preference (F6,49 = 0.26, p = 0.95), latency period until feeding (F6,49 = 0.24, p = 0.96) or home-cage feed consumption (F6,49 = 0.35, p = 0.91) in control animals. The data represent the means ± standard errors of the mean (SEM) from 8 mice per group. ##p < 0.01 versus the vehicle-control group; *p < 0.05 and **p < 0.01 versus the vehicle-CUMS group.
Further behavioural experiments revealed that treatment with oleanolic acid for 3 weeks had no significant effects on sucrose preference (Fig. 1A) and latency period until feeding (Fig. 1B) in naive mice, indicating that the effect of oleanolic acid was limited to stressed animals.
Oleanolic acid restores miR-132 downregulation induced by CUMS
To further investigate the possible mechanisms that underlie the oleanolic acid–induced neurogenic effects, we measured the expression of BDNF, which is important for synaptic protein and neuronal proliferation in the brain. Our results showed that oleanolic acid markedly reversed the BDNF downregulation induced by CUMS (Fig. 2).

Oleanolic acid reverses the change in hippocampal brain-derived neurotrophic factor (BDNF) and miR-132 expression induced by chronic unpredictable mild stress (CUMS). (A) Representative western blot images of BDNF and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (B) Oleanolic acid reversed the decrease in the hippocampal BDNF induced by CUMS (F6,21 = 37.202, p < 0.001). (C) Oleanolic acid reversed the decrease in the hippocampal miR-132 induced by CUMS (F6,21 = 37.766, p < 0.001). The data represent means ± standard errors of the mean (SEM) from 4 mice per group. ##p < 0.01 versus the vehicle-control group; *p < 0.05 and **p < 0.01 versus the vehicle-CUMS group.
In parallel, miR-132 expression, which was significantly inhibited by CUMS, was increased by treatment with both oleanolic acid and fluoxetine.
BDNF-related miR-132 upregulation mediates antidepressant-like effects of oleanolic acid
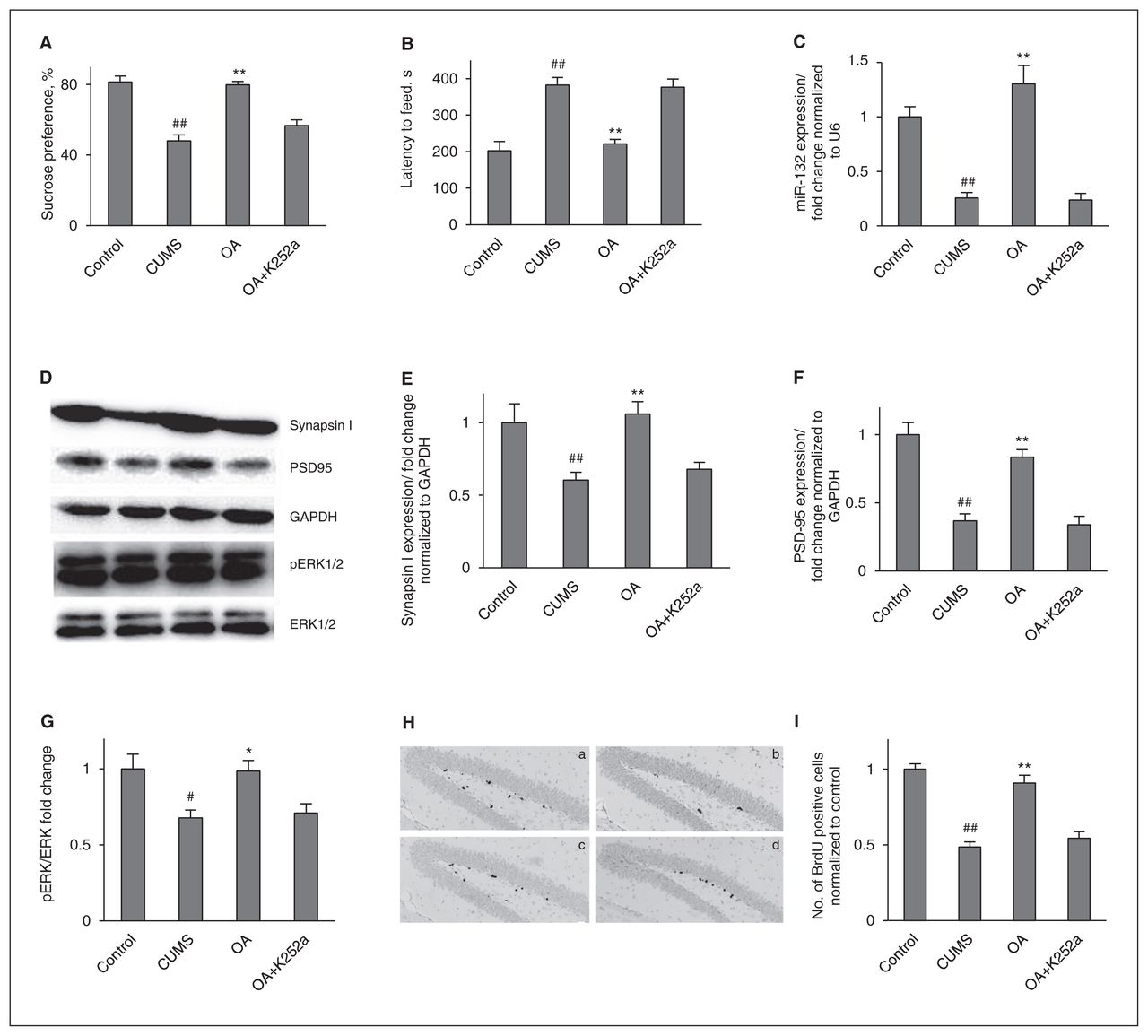
To further determine whether BDNF-TrkB signalling is necessary for the antidepressant-like effects of oleanolic acid, mice in the CUMS groups were cotreated for 3 weeks with oleanolic acid (20 mg/kg) and K252a, a selective inhibitor of the tyrosine protein kinase activity of the trk family of neurotrophin receptors. The behavioural changes were examined 24–48 hours after the last injection. As shown in Figure 3, the K252a injections abolished the antidepressant-like effects of oleanolic acid.

Brain-derived neurotrophic factor (BDNF) is necessary for the behavioural and neurogenic effects of oleanolic acid (OA). (A) K252a suppresses the increase of sucrose preference induced by OA administration. Control versus chronic unpredictable mild stress (CUMS; F1,14 = 49.966, p < 0.001); treatment in the CUMS groups (F2,21 = 31.404, p < 0.001). (B) K252a suppresses the reduction of the latency period until feeding induced by OA administration. Control versus CUMS (F1,14 = 29.533, p < 0.001); treatment in the CUMS groups (F2,21 = 22.429, p < 0.001). (C) The enhancement of miR-132 expression induced by OA administration was blocked by K252a pretreatment. Control versus CUMS (F1,6 = 49.196, p < 0.001); treatment in the CUMS groups (F2,9 = 32.129, p < 0.001). (D) Representative western blot images. (E) K252a abolished the increase of hippocampal synapsin I induced by OA administration. Control versus CUMS (F1,6 = 7.792, p = 0.032); treatment in the CUMS groups (F2,9 = 14.426, p = 0.002). (F) K252a abolished the increase of hippocampal postsynaptic density protein 95 (PSD95) induced by OA administration. Control versus CUMS (F1,6 = 39.782, p = 0.001); treatment in the CUMS groups (F2,9 = 25.015, p < 0.001). (G) K252a abolished the increase of hippocampal p-extracellular signal-regulated kinases (pERK) induced by OA administration. Control versus CUMS (F1,6 = 8.675, p = 0.026); treatment in the CUMS groups (F2,9 = 7.468, p = 0.012). (H) The enhancement of neuronal cell proliferation induced by OA administration was blocked by coinjection with K252a: (a) = control, (b) = CUMS, (c) = CUMS+OA, (d) = CUMS+OA+K252a. (I) Quantification of bromodeoxyuridine (BrdU)+ cells in the hippocampus. Control versus CUMS (F1,4 = 105.75, p = 0.001); treatment in the CUMS group (F2,6 = 27.564, p = 0.001). The data represent the means ± standard errors of the mean (SEM) from 3 or 4 mice per group. GAPDH = glyceraldehyde 3-phosphate dehydrogenase. #p < 0.05 and ##p < 0.01 versus the vehicle-control group; *p < 0.05 and **p < 0.01 versus the vehicle-CUMS group.
Because miR-132 is regulated by BDNF, we next examined whether K252a injection blocked the neurogenic effect of oleanolic acid. To this end, we either administered BrdU injections or sacrificed the mice immediately after the NSFT. We found that K252a pretreatment dramatically prevented the oleanolic acid–induced elevation of miR-132 expression. Moreover, as miR-132 has a positive effect on the increase in neuronal outgrowth and synaptic protein levels, we measured the presynaptic protein synapsin I, postsynaptic protein PSD95 and neuronal cell proliferation. Oleanolic acid significantly increased synapsin I, PSD95 and neuronal cell proliferation. Interestingly, these increases were weakened by K252a injection. Together, these results suggest that BDNF signalling is necessary for the regulation of miR-132 by oleanolic acid.
In addition, since the ERK pathway is a key downstream signalling pathway of BDNF and upstream signalling pathway of miR-132, we also examined the BDNF–TrkB–ERK downstream signalling pathway induced by oleanolic acid in combination with K252a. There was a significant difference in pERK between the control and CUMS groups. Oleanolic acid counteracted the pERK deficiency induced by CUMS. More importantly, this regulation of oleanolic acid was fully blocked by K252a injections.
ERK signalling mediates miR-132 regulation of oleanolic acid
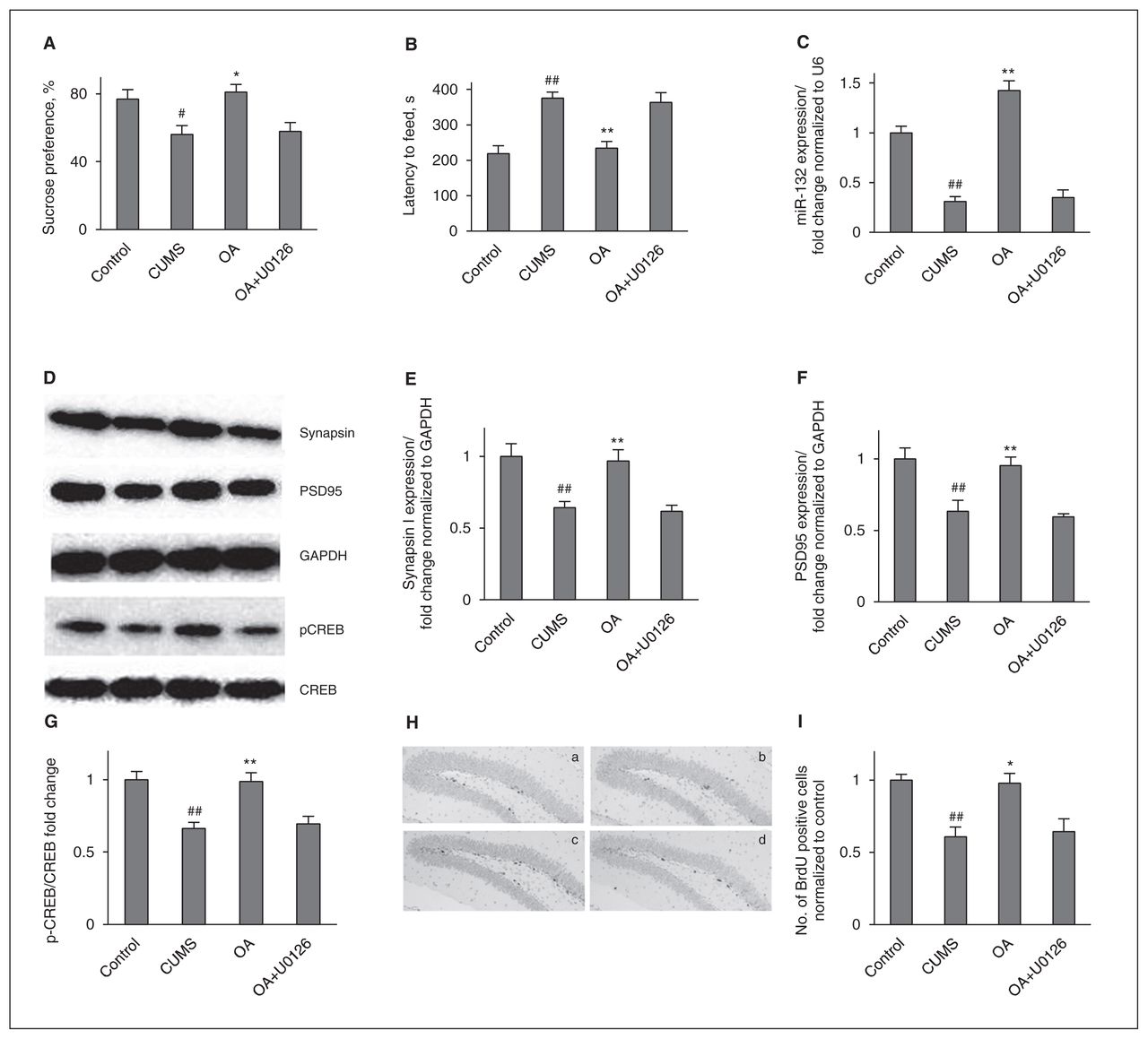
To further investigate the possibility that the ERK pathway is involved in the oleanolic acid–potentiated miR-132 and neuronal proliferation, we examined the effects of U0126 in our system. In the presence of U0126 (0.4 mg/kg/day), the oleanolic acid–induced behavioural improvement was completely blocked (Fig. 4). In addition, we examined the effects of U0126 on the oleanolic acid–induced neurogenic improvement. We found that miR-132, presynaptic protein synapsin I, PSD95 and neuronal cell proliferation were abolished by pretreatment with the inhibitor.

Extracellular signal-regulated kinases (ERK) signalling is necessary for the behavioural and neurogenic effects of oleanolic acid (OA). (A) U0126 suppresses the increase of sucrose preference induced by OA administration. Control versus chronic unpredictable mild stress (CUMS; F1,14 = 7.437, p = 0.016); treatment in the CUMS groups (F2,21 = 7.728, p = 0.003). (B) U0126 suppresses the reduction of the latency period until feeding induced by OA administration. Control versus CUMS (F1,14 = 29.872, p < 0.001); treatment in the CUMS groups (F2,21 = 12.688, p < 0.001). (C) The enhancement of miR-132 expression induced by OA administration was blocked by U0126 pretreatment. Control versus CUMS (F1,6 = 68.61, p < 0.001); treatment in the CUMS groups (F2,9 = 67.096, p < 0.001). (D) Representative western blot images. (E) K252a abolished the increase of hippocampal synapsin I induced by OA administration. Control versus CUMS (F1,6 = 13.236, p = 0.011); treatment in the CUMS groups (F2,9 = 11.531, p = 0.003). (F) K252a abolished the increase of hippocampal postsynaptic density protein 95 (PSD95) induced by OA administration. Control versus CUMS (F1,6 = 11.171, p = 0.016); treatment in the CUMS groups (F2,9 = 11.312, p = 0.003). (G) K252a abolished the increase of hippocampal p-cyclic adenosine monophosphate response element binding protein (pCREB) induced by OA administration. Control versus CUMS (F1,6 = 23.296, p = 0.003); treatment in the CUMS groups (F2,9 = 11.984, p = 0.003). (H) The enhancement of neuronal cell proliferation induced by OA administration was blocked by coinjection with U0126: (a) = control, (b) = CUMS, (c) = CUMS+OA, (d) = CUMS+OA+U0126. (I) Quantification of bromodeoxyuridine (BrdU)+ cells in the hippocampus. Control versus CUMS (F1,4 = 25.00, p = 0.007); treatment in the CUMS groups (F2,6 = 7.628, p = 0.022). The data represent means ± standard errors of the mean (SEM) from 3 or 4 mice per group. GAPDH = glyceraldehyde 3-phosphate dehydrogenase. #p < 0.05 and ##p < 0.01 versus the vehicle-control group; *p < 0.05 and **p < 0.01 versus the vehicle-CUMS group.
Next, because miR-132 is regulated by CREB, we also evaluated the CREB levels after oleanolic acid treatment or cotreatment with U0126. We found reduced CREB phosphorylation in the CUMS groups compared with the control groups. Chronic oleanolic acid administration totally counteracted this stress-induced abnormality. This role of oleanolic acid was fully blocked by U0126 coinjections. These results suggest that ERK-CREB is required for the oleanolic acid–potentiated miR-132.
miR-132 expression is necessary for neuronal cell proliferation by oleanolic acid
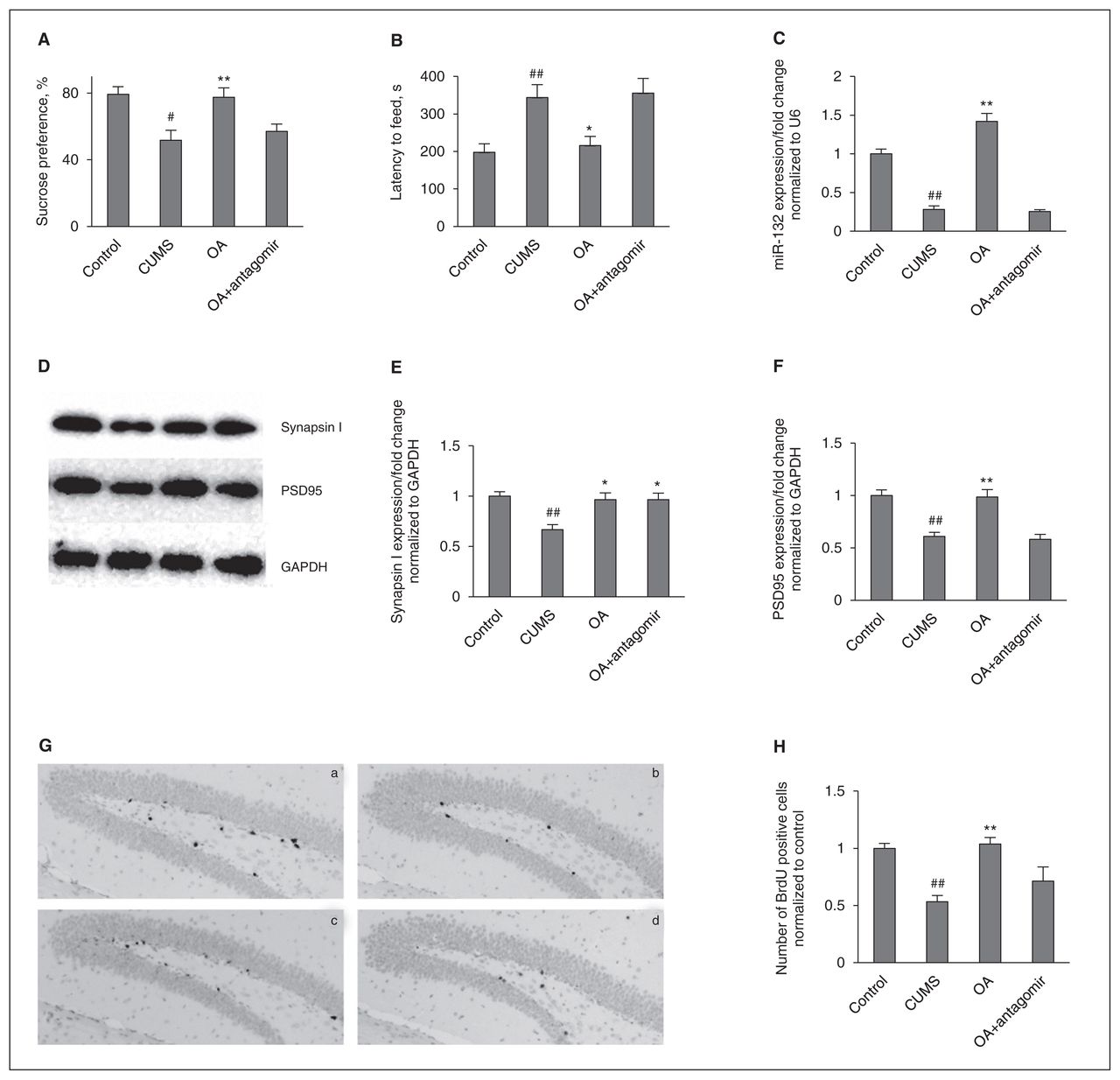
To verify the possible involvement of miR-132 in the regulation of synaptic protein expression and neuronal cell proliferation, we examined the effect of an antagomir to inhibit the miR-132 function. Treatment with an miR-132 antagomir significantly reduced the expression of miR-132 in the hippocampus induced by oleanolic acid compared with the miR-132 antagomir control and the oleanolic acid cotreated groups (Fig. 5). Similarly, the miR-132 antagomir also abolished the effect of oleanolic acid on behaviours, neuronal cell proliferation and postsynaptic protein PSD95. However, as shown in Fig. 5E, in contrast to postsynaptic protein PSD95, no change in the levels of the presynaptic protein synapsin I was caused by the miR-132 antagomir with or without oleanolic acid.

miR-132 expression is necessary for the behavioural and neurogenic effects of oleanolic acid (OA). (A) The miR-132 antagomir suppresses the increase of sucrose preference induced by OA administration. Control versus chronic unpredictable mild stress (CUMS; F1,14 = 13.306, p = 0.003); treatment in the CUMS groups (F2,21 = 6.402, p = 0.007). (B) The miR-132 antagomir suppresses the reduction of the latency period until feeding induced by OA adminstration. Control versus CUMS (F1,14 = 12.856, p = 0.003); treatment in the CUMS group (F2,21 = 5.466, p = 0.012). (C) The enhancement of miR-132 expression induced by OA administration was blocked by miR-132 antagomir pretreatment. Control versus CUMS (F1,6 = 92.376, p < 0.001); treatment in the CUMS groups (F2,9 = 105.251, p < 0.001). (D) Representative western blot images. (E) The miR-132 antagomir did not affect the increase of hippocampal synapsin I induced by OA administration. Control versus CUMS (F1,6 = 25.379, p = 0.002); treatment in the CUMS groups (F2,9 = 7.637, p = 0.012). (F) The miR-132 antagomir abolished the increase of hippocampal postsynaptic density protein 95 (PSD95) induced by OA administration. Control versus CUMS (F1,6 = 36.071, p = 0.001); treatment in the CUMS groups (F2,9 = 18.027, p = 0.001). (G) The enhancement of neuronal cell proliferation induced by OA administration was blocked by coinjection with the miR-132 antagomir: (a) = control, (b) = CUMS, (c) = CUMS+OA+antagomir-control, (d) = CUMS+OA+miR-132 antagomir. (H) Quantification of bromodeoxyuridine (BrdU)+ cells in the hippocampus. Control versus CUMS (F1,4 = 44.576, p = 0.003); treatment in the CUMS groups (F2,6 = 9.259, p = 0.015). The data represent means ± standard errors of the mean (SEM) from 3 or 4 mice per group. GAPDH = glyceraldehyde 3-phosphate dehydrogenase. #p < 0.05 and ##p < 0.01 versus the vehicle-control group; *p < 0.05 and **p < 0.01 versus the vehicle-CUMS group.
Discussion
The present study provides in vivo evidence that silencing miR-132 in mice using antagomirs prevents behavioural improvement and neurotrophic signalling induced by oleanolic acid and shows that oleanolic acid administration prevents chronic stress-impaired neuronal proliferation through TrkB receptor–mediated modulation of hippocampus BDNF–ERK–CREB–miR-132 signalling. To our knowledge, this study is the first to analyze miR-132 expression in a depression-like model of CUMS.
Oleanolic acid is a natural pentacyclic triterpene that is widely distributed in food and medicinal plants. It can be found in Prunus mume, Vitis vinifera, Sambucus chinesis, Panax ginseng, Oldenlandia diffusa and Rabdosia rubescens.31–33 We recently reported that oleanolic acid increased BDNF levels and produced antidepressant behavioural effects in stressed animals.19 In the present study, we used another efficacious CUMS procedure to mimic stress in daily life in animals. A decreased sucrose preference and a prolonged latency to feed induced by the CUMS procedure were significantly restored by a 3-week regimen of oleanolic acid administration, which confirmed the antidepressant-like effect of oleanolic acid. In addition to behavioural deficits, one widespread feature of major depression is the inhibition of neurotrophic system regulation.3 Consistent with the results of our previous study, the present results also indicated that oleanolic acid ameliorated the deficits in hippocampal BDNF expression induced by CUMS.19
Within the central nervous system, microRNAs have emerged as important effectors of an array of developmental, physiologic, and cognitive processes.34 We focused on the enhancement of BDNF-induced miR-132 by treatment with oleanolic acid because miR-132 can promote neural growth,35 synaptic function16,36 and cognitive deficits,37 which were associated with antidepressant action. In addition, miR-132 was significantly upregulated in nonlearned, helpless rats compared with learned helpless ones.38 We therefore detected this microRNA expression in a depression-like model of CUMS. The data provided from PCR indicated that CUMS suppressed the levels of miR-132 in the hippocampus. Previous studies have shown that BDNF upregulated the levels of miR-132 in cortical cultures and dopamine neurons,39,40 that miR-132 overexpression enhanced neurite outgrowth in PC12 cells35 and that miR-132 knockdown impaired the integration of newborn neurons into the excitatory synaptic circuitry of the adult brain.41 Moreover, decreased expression of miR-132 after glucocorticoid treatment in vitro has also been reported.16 Chronic constriction injury steadily decreased the relative expression of miR-132 in the rat hippocampus.42 Thus, findings on this particular miRNA using CUMS may be extrapolated to other depression models. On the other hand, the finding that miR-132 was restored by fluoxetine and oleanolic acid suggests that miR-132 may serve as a hippocampal target of antidepressant treatments. In addition, the finding of decreased PSD95 expression and neuronal cell proliferation resulting from CUMS is consistent with the findings of a previous study demonstrating that chronic stress causes dendritic atrophy of neurons in the brain.43 Oleanolic acid administration for 3 weeks significantly attenuated these deficits.
The present work also showed that the CUMS-induced reductions of sucrose preference and BDNF expression were attenuated by chronic fluoxetine treatment, which is consistent with reults of a previous study.7 At the same time, fluoxetine also upregulated miR-132 expression in hippocampus. Previous researches have demonstrated that fluoxetine possessed the ability to increase miR-598 and miR-451 expression in the rat hippocampus44 and down-regulate miR-16 levels in the mouse hippocampus.45 Our finding supplies additional evidence to demonstrate the involvement of hippocampal miR-132 in fluoxetine treatment. In addition, it should be noted that treatment with oleanolic acid and fluoxetine elevated miR-132 above control levels. In fact, the increase of miR-132 outweighed that of BDNF during neuronal differentiation.40 In addition, the expression of miR-132 increased up to 4-fold the sensitivity induced by BDNF.39 Some potent antidepressant agents, such as luteolin increased miR-132 levels 2-fold above control levels. We speculate that this elevation may be attributed to the additional elevation of BDNF levels by oleanolic acid and fluoxetine treatments.
In an attempt to move beyond receptor/ligand-based theories of pathology, signalling transduction pathways have become a new focus in depression research.46 The BDNF–TrkB–ERK downstream signalling pathway has been proposed as an intracellular signalling mechanism mediating antidepressant efficacy and miR-132 regulation.14 Therefore, mice were coinjected with oleanolic acid (20 mg/kg) and K252a (25 μg/kg), a potent pharmacological inhibitor of the BDNF receptor TrkB, for 3 weeks. The results indicated that K252a abolished the antidepressant-like effects of oleanolic acid in the SPT and NSFT and inhibited hippocampal miR-132, synapsin I, PSD95 and neuronal cell proliferation. Combined with previous findings that upregulation of miR-132 was caused by BNDF,16 these results confirmed that oleanolic acid, through BDNF–miR-132, positively regulated synaptic protein levels and neural growth.
As the activation of p-ERK induced by oleanolic acid was suppressed by K252a, to further investigate the possibility that the ERK pathway is involved in the oleanolic acid– potentiated miR-132 and neuronal proliferation, we examined the effects of U0126, an ERK inhibitor, in our system. In addition to miR-132, we also examined the changes in CREB activity, because CREB is a downstream regulator of the ERK cascade and CREB phosphorylation has also been proposed as an intracellular signalling mechanism that mediates neuron proliferation and cognitive deficits.20 On the other hand, CREB could regulate the transcription of miR-132.12 The effect of oleanolic acid on CREB phosphorylation was abolished using the ERK inhibitor U0126, and it inhibited miR-132, PSD95 expression and neuronal cell proliferation, suggesting that the hippocampal ERK–CREB signalling pathway is necessary for upregulation of miR-132 and the antidepressant-like effect of oleanolic acid. Consistent with our study, a recent study also demonstrated that the transcription of miR-132 is strongly regulated by the ERK signalling pathway in neurons.14 Therefore, the BDNF–ERK–CREB signalling pathway is involved in the antidepressant-like effect of oleanolic acid and regulation of miR-132.
We next used antagomirs to reduce hippocampal miR-132 levels, thereby modelling the oleanolic acid-induced miR-132 profile. Antagomirs are known to function by complementary binding, leading to non–RNAi-mediated degradation of the miRNA.47 Because a previous study confirmed that antagomirs efficiently silence target miRNAs in the central nervous system when they are only injected locally into the mouse brain,48 the miR-132 antagomir was infused into the lateral ventricle in the present study. The results demonstrated that miR-132 antagomir profoundly abolished oleanolic acid–induced neuronal cell proliferation. Very recently, Luikart and colleagues41 reported that knockdown of miR-132 impaired the integration of newborn neurons, a bio-marker that can be enhanced by antidepressants.49 Together, our findings suggest that miR-132 plays an important role in the effects of oleanolic acid on neuronal growth in vivo.
Interestingly, we found that antagomirs targeting miR-132 attenuated only oleanolic acid–induced postsynaptic protein PSD95. On the contrary, presynaptic protein synapsin I was not influenced by the miR-132 antagomir. Previous studies also suggest a model in which enhanced levels of BDNF lead to changes in the neuronal transcriptome directly via the repression of target gene expression and indirectly via the regulation of neuronal miRNA expression.50,51 In addition, Kawashima and colleagues16 found that the transfection of antisense RNA to inhibit miR-132 function decreased the BDNF-dependent increase in the expression of postsynaptic proteins, but did not affect the expression of presynaptic proteins. Moreover, considering that ERK is able to regulate transcription both by the direct phosphorylation of specific transcription factors and by the indirect activation of downstream kinases or miRNAs,14 it is possible that oleanolic acid regulates postsynaptic protein via miR-132-dependent mechanisms and regulates presynaptic protein via miR-132- independent mechanisms.
Limitations
It should be noted that our study did not have a vehicle control group with K252a or U0126, thus an antagonist or inhibitor effect may have contributed to our results. This is one of the limitations of the present study. In our previous study, 25 μg/kg of K252a did not alter animal behaviours.22 In addition, in our preliminary study, neither sucrose preference nor latency period until feeding was affected by U0126 in mice exposed to CUMS. Moreover, previous studies also found that treatment with U0126 alone did not induce any alteration in miR-132 expression.35,39 A control experiment in a future study will be helpful to show that an antagonist or inhibitor at the dose tested does not induced related actions.
It is generally accepted that several other stimuli can also induce CREB phosphorylation in the hippocampus. Thus, regulation of miR-132 may not be restricted to neurotrophic signalling. Another signalling pathway, such as protein kinase A, could have contributed to miR-132 regulation. In addition, it is not entirely clear how miR-132 levels could be used as a hippocampal target of antidepressant treatments, as other depression-like models and clinical antidepressants were not evaluated in the present study.
Conclusion
Our results provide direct evidence that oleanolic acid exerts antidepressant-like properties in an animal model of depression, and these effects appear to be partly mediated through upregulation of the hippocampal BDNF–TrkB–ERK–CREB–miR-132 signalling pathway. In addition, we have demonstrated that miR-132 is inhibited in CUMS-induced depression, and it is critical for the antidepressant-like effect of oleanolic acid. Our study provides a future direction for the examination of mechanisms of depression related to miR-132 as well as further insight into the possible therapeutic use of oleanolic acid for treating major depression.
Acknowledgements
The project was supported by grants from the National Natural Science Foundation of China (No. 81202940), and the Science Research Foundation of ministry of Health & United Fujian Provincial Health and Education Project for Tacking the Key Research (WKJ-FJ-31).
Footnotes
Competing interests: None declared.
Contributors: L.-T. Yi designed the study. J. Li, B.-B. Liu and L. Luo carried out the experiment. B.-B. Liu, L. Luo and D. Geng acquired the data, which J. Li and Q. Liu analyzed. L.-T. Yi wrote the article, which all other authors reviewed. All authors approved publication.
- Received August 8, 2013.
- Revision received January 14, 2014.
- Revision received March 3, 2014.
- Revision received March 17, 2014.
- Revision received March 25, 2014.
- Accepted March 28, 2014.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.