

Abstract
Major depression and anxiety are highly prevalent and involve chronic dysregulation of serotonin, but they remain poorly understood. Here, we review novel transcriptional (genetic, epigenetic) and posttranscriptional (microRNA, alternative splicing) mechanisms implicated in mental illness, focusing on a key serotonin-related regulator, the serotonin 1A (5-HT1A) receptor. Functional single-nucleotide polymorphisms and stress-induced DNA methylation of the 5-HT1A promoter converge to differentially alter pre- and postsynaptic 5-HT1A receptor expression associated with major depression and reduced therapeutic response to serotonergic antidepressants. Major depression is also associated with altered levels of splice factors and microRNA, posttranscriptional mechanisms that regulate RNA stability. The human 5-HT1A 3′-untranslated region is alternatively spliced, removing microRNA sites and increasing 5-HT1A expression, which is reduced in major depression and may be genotype-dependent. Thus, the 5-HT1A receptor gene illustrates the convergence of genetic, epigenetic and posttranscriptional mechanisms in gene expression, neurodevelopment and neuroplasticity, and major depression. Understanding gene regulatory mechanisms could enhance the detection, categorization and personalized treatment of major depression.
Introduction
Major depression is a serious illness, with 15% lifetime prevalence; it is ranked number 1 in terms of global burden of disease. 1 Both genetic and environmental factors are thought to contribute to major depression. Anxiety and depression are closely related genetically,2 and are often comorbid. They are thought to involve reduced serotonin (5-HT) activity,3–6 which is reversed by selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine.7–11 However, treatment with SSRIs for 5 to 8 weeks is required for remission, which occurs in only 30% of patients.12–14 Thus, although the serotonin system is involved in major depression and antidepressant actions, a better understanding of how the system is regulated over time is needed to further enhance the effectiveness of treatment.8,9
The brain 5-HT system originates in the raphe nuclei, which projects widely15 to innervate corticolimbic systems involved in stress,16 anxiety,17 depression18 and cognitive function. 19 The levels of 5-HT are determined primarily by expression of the rate-limiting enzyme tryptophan hydroxylase-2 (TPH2);20 by reuptake via the 5-HT transporter (5-HTT), which is the target of SSRI antidepressants;21 and by the levels of 5-HT1A autoreceptors that negatively regulate 5-HT neuronal firing.22 Increased levels of 5-HT1A autoreceptors, which would reduce 5-HT activity, are seen in depressed patients and in the raphe tissue of people with depression who died by suicide (Fig. 1).23,24 By blocking the 5-HTT, SSRIs allow released 5-HT to remain in the synaptic space and persistently activate receptors on target neurons.25 However, 5-HT is also augmented in the raphe, which activates the 5-HT1A autoreceptor and mediates feedback inhibition to reduce 5-HT neuron firing, negating the effect of SSRI treatment.25 But after days to weeks of SSRI treatment, the 5-HT1A autoreceptor desensitizes.26,27 Although acute desensitization mechanisms such as uncoupling and internalization are occurring, 28–30 the long time course suggests a role for transcriptional regulation.31 Altered regulation of the genes for 5-HTT and 5-HT1A receptors could alter 5-HT activity, predispose to depression and affect SSRI response.
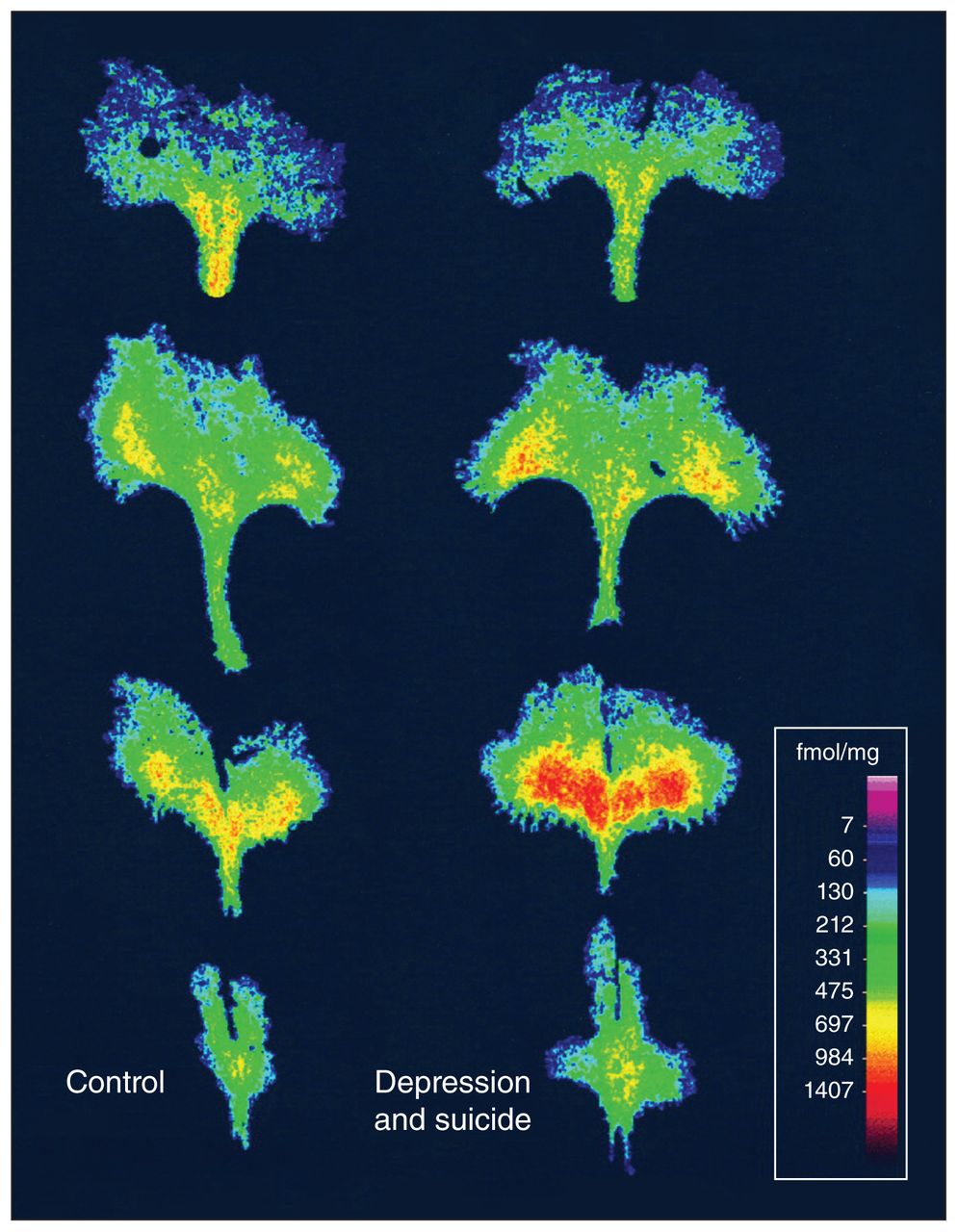
Increased levels of 5-HT1A autoreceptors in the dorsal raphe of depressed individuals who died by suicide compared with the brains of healthy people. Digitized images of [3H]DPAT binding to 5-HT1A receptors at 4 rostral-to-caudal levels of the dorsal raphe from a representative control participant (left) and an age-matched person who had major depression and died by suicide (right). Reproduced from Stockmeier and colleagues24 with permission from Journal of Neuroscience.
In this review, we discuss how different transcriptional and posttranscriptional mechanisms alter the regulation of these key genes in depression, focusing on the 5-HT1A receptor gene (Fig. 2). The 5-HT1A receptor gene provides an example of how global alterations in these regulatory mechanisms may affect the expression of many genes. We present evidence from human and animal model studies that link multiple regulatory changes with increased risk of depression, anxiety and resistance to SSRI treatment.

Model of key transcriptional, epigenetic and posttranscriptional regulatory sites of the human HTR1A gene. The promoter, coding sequence and 3′-untranslated region of the 5-HT1A receptor gene are shown, highlighting key regulatory regions altered in major depression by genotype (SNPs rs6295 and rs878567), DNA methylation (Sp4 site), microRNA (miR-135), and alternative splicing. Shown are the transcription start (arrow) and polyadenylation (A) sites, DNA-binding transcription factors (DEAF1, MeCP2, Sp4) and RNA-binding splice factors (PTBP1, nSR100, RBFOX1) implicated. Activation (arrow) or inhibition (block) may be dependent (dashed line) on genotype, methylation or splicing. 5-HT = serotonin; CDS = coding sequence; SNP = single nucleotide polymorphism.
Genetic risk: transcriptional mechanisms
A consistent finding in genome-wide association studies has been that single-nucleotide polymorphisms (SNPs) associated with depression are localized in noncoding and often gene-regulatory or untranslated regions.2 Another consistent finding has been the overlap of genes associated with different mental illnesses, the greatest similarity being between depression and anxiety.2 This implicates altered transcriptional regulation as a potential primary change that confers genetic risk for depression and related mental illnesses.
Functional 5-HT polymorphisms and major depression
In the 5-HT system, the 5-HTTLPR (long polymorphic repeat) located in the promoter region of the 5-HTT gene was the first to be associated with anxiety-like behaviour and depression. 32,33 The 5-HTTLPR ss genotype reduces 5-HTT transcription in vitro34 and has been associated with reduced 5-HTT expression in blood cells,32 although this association has not been detected in the brain.35 The 5-HTT ss genotype has been associated with major depression and resistance to SSRI treatment in many studies,36 but not all.37 Subsequently, the 5-HT1A rs6295 SNP was identified,38 and the GG genotype was shown to lead to overexpression of 5-HT1A autoreceptors by preventing binding of the repressors DEAF1/NUDR and HES5/HES1 (Fig. 2). Because DEAF1 functions as an HTR1A repressor in raphe cells, but as an enhancer in postsynaptic neuronal cells,39 the rs6295 G allele should increase 5-HT1A autoreceptor expression, but reduce postsynaptic 5-HT1A heteroreceptor levels. In support of this, recent studies have shown an association of the rs6295 GG genotype with reduced 5-HT1A receptors in cortical and hippocampal tissues,40,41 while there is increased 5-HT1A binding in the raphe.23,41 Similarly, knockout of Deaf1 in mice induces raphe 5-HT1A autoreceptors while reducing prefrontal cortical 5-HT1A expression,42 implicating DEAF1 in the effects of the rs6295 polymorphism, although other factors may also participate.40 Thus, rs6295 is a functional promoter polymorphism that modifies 5-HT1A receptor gene expression in a region-specific manner.
In previous reviews, we presented evidence that, similar to the 5-HTTLPR, the HTR1A rs6295 risk genotype (G allele or GG genotype) has been associated with major depression and resistance to the antidepressant actions of SSRIs and atypical antipsychotics in multiple studies.43,44 Associations of rs6295 with major and bipolar depression are supported by meta-analyses, 45–47 although no association with anxiety or depression symptoms was reported in a large cohort of healthy people. 48 Furthermore, several other 5-HT-, monoamine-, or stress-related genes have also been associated with major depression. 49,50
The HTR1A G carrier or GG genotype has also been associated with increased 5-HT1A autoreceptors,23,41 major depression, 51 amygdala reactivity to fearful stimuli52–56 and increased hippocampal volume.57 In addition, associations have been reported between the G carrier or the GG genotype and disordered eating in female adolescents,58 panic disorder59 or panic disorder without agoraphobia,60 substance abuse and psychiatric hospitalization.61 Depressed patients with the C allele showed enhanced response to transcranial magnetic stimulation,62 similar to the association with SSRI response. The rs6295 polymorphism was associated with SSRI-induced adverse effects: diarrhea (G allele) and sexual apathy (C allele).63 One study found that the rs6295 G allele was associated with greater (rather than reduced) response to SSRI treatment.64 Therefore, it was important to test in animal models whether altered transcription of 5-HT1A autoreceptors or heteroreceptors has any influence on behaviour or response to antidepressants.
Genetic interactions
Several genetic interactions of rs6295 with risk alleles, such as 5-HTTLPR,65 HTR2A rs6311, or BDNF rs6265 SNPs in major depression, have been reported.66 The HTR1A rs6295 polymorphism showed strong interaction with life stress and BDNF rs6265 (Val66Met) for major depression67 and for treatment-resistant depression.68 The BDNF Val66Met has the highest level of functionality (class 369), having been shown to phenocopy reduced BDNF trafficking and secretion in a mouse knockin model.70 In the human brain, the BDNF Met allele was associated with reduced 5-HT1A binding in 1 study,71 but not others.72,73 In a recent study, 3 or more combined HTR1A and BDNF risk alleles associated increase 5-HT1A receptor binding in cortical subregions.74 In contrast, the COMT Val158Met rs4680 genotype oppositely altered the rs6295 association with interferon-induced depression.75 Perhaps most intriguing is an interaction of the HTR1A rs6295 genotype with the phospholysine phosphohistidine inorganic pyrophosphate phosphatase (LHPP) gene in major depression, seen in Utah and Ashkenazi populations. Subsequently, LHPP was identified in a genome-wide association study of female melancholic depression76 and a Mexican–American major depression cohort.77 Recently, LHPP has been shown to function as a tumour suppressor,78 and it may affect 5-HT1A receptor signalling, but this remains to be addressed.79
HTR1A rs6295 and phenotypic/brain network alterations
In terms of physiologic differences, rs6295 G carriers showed thermal hypoalgesia80 and increased depression after lumbar surgery.81 In panic patients with disorder/agoraphobia with the GG genotype, there was increased amygdala reactivity to threat or safety cues, behavioural avoidance and reduced response to cognitive behavioural therapy,82 and increased contextual fear in healthy participants.83 Similarly, in panic disorder the G allele was associated with increased amygdala reactivity to fearful faces,56 while in healthy controls an opposite association was seen,52 suggesting that the influence of the polymorphism may depend on the disease state.22 In small studies of the elderly, the C allele was protective against depressed mood in healthy athletes compared to nonathletes (n = 55 v. 58)84 and enhanced remission to 4-week citalopram treatment in people recently diagnosed with depression (n = 19).85 In addition, the G allele was inversely correlated with parasympathetic tone, which was associated with reduced anxiety in healthy participants (n = 1141).86 Thus, the rs6295 G allele associates with depression and anxiety phenotypes, while the C allele may be protective.
The HTR1A rs6295 genotype also associates with alterations in brain activity that may underlie depression. Brain imaging studies of people with bipolar disorder in the depressed state have shown that the GG genotype is associated with increased amygdala–ventrolateral prefrontal cortex (PFC) connectivity, and changes in corticolimbic connectivity correlated with depression severity.87 In a study of 99 healthy Japanese people, the G carriers were associated with decreased functional connectivity of the default mode network (DMN) in the dorsolateral and ventromedial PFC, and with reduced salience network connectivity in the ventromedial PFC and the subgenual anterior cingulate cortex.88 Abnormalities in these networks, particularly the DMN, have been associated with major depression, and with the 5-HTTLPR ss genotype,89,90 as well as with other 5-HT-, monoamine- or neuropeptide-related genes.91 Other studies have shown that levels of raphe and cortical 5-HT1A receptors detected using [11C]WAY100635 binding are increased in depressed patients and correlate with cortical thickness and tract number in the posterior cingulate cortex, part of the DMN.92 Conversely, in healthy people [11C]WAY100635 binding was negatively correlated with posterior cingulate cortex and medial dorsal PFC activity.93 These studies suggest that the 5-HT1A receptor regulates the DMN, but in opposite ways in healthy people compared to people with depression. Increased activity of the DMN could correlate with the increased rumination and self-focus observed in major depression.94
Cognitive function was also affected in people with the G allele, with poorer performance on the Iowa gambling test (in combination with 5-HTTLPR ss)95 and impaired working memory in premenstrual dysphoric disorder.96 Conversely, depressed patients with the CC genotype showed better performance in a battery of 9 cognitive tests.97 In healthy Han Chinese people, the CG/GG genotype has been linked with difficulty to identify one’s own feelings and in forming close attachments98 or romantic relationships, 99 which could relate to depression susceptibility. In panic disorder, the CC genotype is associated with increased fractional anisotropy of the left cingulate gyrus.100 In addition, 5-HT1A receptors ([18F]-FCWAY binding sites) are reduced in the anterior cingulate cortex in panic disorder, 101 and the GG genotype is associated with augmented avoidance and reduced enhancement of anterior cingulate cortex activity by therapy.82 These imaging findings suggest that genotype-induced changes in 5-HT1A receptor expression affect the structure of brain networks implicated in mood and cognitive function, particularly in anxiety.
Genetic risk: animal models
To test the importance of transcriptional mechanisms in the effects of gene polymorphisms, animal models have been used.69 Ideally, knockin of the specific polymorphism provides an unequivocal validation of the behavioural effect of that polymorphism (functional phenocopy), as has been done for the BDNF Val66Met (rs6265) polymorphism.70
HTR1A rodent models
Several mouse models support the role of DEAF1 and 5-HT1A autoreceptors in regulation of the 5-HT system and in major depression and anxiety. Because the sequence of the DEAF1 site is not highly conserved in mouse and human HTR1A genes and mice lack the rs6295 polymorphism,42 a mouse knockin model of the rs6295 SNP may not be functional. Furthermore, the mouse HTR1A gene has 2 DEAF1 sites, one of which is critical for DEAF1 repression, and the other seems to also mediate DEAF1 enhancer activity.102 Nevertheless, insertion of the human HTR1A gene (C and G alleles) in a 5-HT1A knockout mouse background indicates region- and allele-specific differences in HTR1A expression, despite restricted HTR1A expression in this model.103 Global knockout of DEAF1 leads to increased 5-HT1A autoreceptor RNA and protein levels in the raphe, while the PFC had reduced 5-HT1A receptor expression,42 consistent with PET imaging results in depressed patients41 and in vitro studies.39 However, the DEAF1 knockout mice showed a sex- and test-dependent anxiety phenotype that was more pronounced in males.104 However, the phenotype was very mild, likely due to a desensitization of G-protein coupling to potassium channels.104 Specific, inducible knockdown in adult male mice of 5-HT1A autoreceptors by 30% resulted in a stress-resilience phenotype with enhanced and more rapid response to SSRI treatment.105 Similarly, acute small interfering RNA (siRNA)–induced knockdown of 5-HT1A autoreceptors in male mice induced a rapid antidepressant-like phenotype within days106 and enhanced response to SSRIs.107 Surprisingly, induced knockout of 5-HT1A autoreceptors in adult mice (both sexes) resulted in hyperactivation of 5-HT neurotransmission and a paradoxical anxiety response to subacute (9-d) SSRI treatment. 108 Hyperactivation of the 5-HT system may relate to suicidality seen in adolescent patients upon SSRI treatment. 109–111 In contrast, deletion of the repressor CC2D1A/Freud-1 in 5-HT neurons of adult mice (both sexes) led to an upregulation of 5-HT1A autoreceptors, and an SSRI-resistant anxiety and depression-like phenotype.112 Importantly, these effects were dependent on increased 5-HT1A autoreceptors and were not seen in a 5-HT1A autoreceptor-deficient background.113 Like the Freud-1 knockout mice, knockout in adult 5-HT neurons of MeCP2, an X-linked methyl-binding protein that interacts with and augments DEAF1 activity, induced 5-HT1A autoreceptors and led to a sex-dependent behavioural phenotype: females had reduced anxiety, whereas males showed increased anxiety and reduced depression-like behaviours.102 Analogous to the Freud-1 knockout model, behavioural response to atypical antipsychotics in an NMDA hypofunction model of schizophrenia was abolished in Pet1 knockout mice, which are deficient in 5-HT neurons.114 Thus, normal function of 5-HT neurons is critical for response to both SSRIs and atypical antipsychotics. These studies suggest that the rs6295-induced upregulation of 5-HT1A autoreceptors drives the increased risk for anxiety- and depression-related phenotypes and confers resistance to SSRI treatment.
HTR1A genotype, neurodevelopment and neuroplasticity
Since genotype is present throughout life, the HTR1A genotype is likely to exert effects on 5-HT1A transcription throughout development, as indicated by a recent study of HTR1A allelic imbalance.61 In postmortem PFC tissue from healthy controls who were heterozygous for the rs6295 polymorphism, the C allele was associated with increased levels of the corresponding 5-HT1A RNA (rs878567 C allele), both in adult and in fetal (from gestational week 18) tissues.61 This indicates that the rs6295 polymorphism influences 5-HT1A transcription throughout life. Several studies have addressed the actions of 5-HT and 5-HT1A receptor dysregulation on behaviour and neurodevelopment using rodent models.115 In 5-HT1A knockout male mice, conditional rescue of forebrain 5-HT1A heteroreceptors during the early postnatal period, but not in adulthood, rescued the anxiety phenotype of these mice.116 In contrast, suppression of forebrain 5-HT1A heteroreceptors in adolescent male mice (p35–p50), but not adult male mice, resulted in adulthood depression-like behaviour.117 On the other hand, lifelong 5-HT1A autoreceptor knockout, or knockdown by 40% in juvenile (p14–p30) male mice, but not adult mice,105,108 also resulted in adulthood anxiety.118,119 During these critical periods, 5-HT innervation to cortical and limbic targets is continuing to mature120,121 and can influence neuroblast migration in these regions.122 Although the role of 5-HT1A receptors in 5-HT neuronal development remains to be elucidated, there is evidence that hippocampal 5-HT1A heteroreceptors signal to CREB to enhance developmental hippocampal synapse formation.123,124 A similar developmental mechanism could be linked to the above-mentioned correlations between 5-HT1A receptor levels and grey-matter thickness in certain brain regions,92 which is altered in major depression.125
In addition to influencing neurodevelopment, there is evidence that antidepressant-induced signalling through 5-HT1A receptors can affect neuroplasticity in adulthood. Chronic SSRI-induced anti-anxiety, antidepressant and hippocampal neurogenesis actions in male mice are dependent on 5-HT1A heteroreceptors expressed on hippocampal granule cells of the dentate gyrus.126 The adult-born hippocampal neurons integrate into the dentate gyrus circuitry to inhibit the activity of mature granule cells, conferring stress resilience.127 There is also evidence that 5-HT1A receptors modulate cortical neuroplasticity in adulthood. For example, in adult rats, chronic fluoxetine induces a rejuvenation of cortical neuroplasticity for ocular dominance, and this effect is mediated by cortical 5-HT1A receptors, signalling through BDNF and ERK1/2.128 Thus, even in adulthood, chronic treatment with SSRIs may influence cortical neuroplasticity via 5-HT1A signalling. In this regard, PFC 5-HT1A receptor signalling to phosphatidyl-inositol 3′-kinase, implicated in the acute antidepressant actions of ketamine in mice,129 is thought to trigger synaptic remodelling.130 The 5-HT1A signalling mechanisms to neuroplasticity-induced behavioural changes remain incompletely understood, as reviewed recently.131
Environmental risk: stress × genotype interaction
In combination with the potential roles of genetic risk factors, a key role for environmental risk factors in mental illness has been shown.66 The Caspi study132 highlighted the cooperative interaction between risk genotype (5-HT transporter long polymorphic repeat, 5-HTTLPR) and environment (number of life stress events) associated with major depression, which has been replicated in many133–136 but not all subsequent studies, 137,138 while a recent study suggests that they are separate, noninteracting risk factors.139 The 5-HTTLPR risk allele (short, s) or genotype (ss) has been associated with altered hippocampal volume57 and amygdala–cingulate cortex reactivity to fear-related stimuli140,141 that can affect response to environmental stress and depression susceptibility. Similarly, work-related stress in a nursing cohort, and major depression in a Japanese cohort were associated with 5-HTT promoter DNA methylation in blood, but no interaction was seen with 5-HTTLPR genotype.142,143 However, in poststroke depression an interaction with increased 5-HTT promoter methylation was seen only in people with the 5-HTTLPR ss genotype.144 Thus, genotype and stress-induced DNA methylation may interact at specific risk gene promoters to predispose people to major depression.67
Stress × HTR1A genotype interaction
Like 5-HTTLPR, there is evidence that the HTR1A promoter polymorphism interacts with stress for susceptibility to anxiety- and depression-related phenotypes. For example, life separation events interacted with both 5-HTTLPR and HTR1A rs6295 polymorphisms in panic disorder.145 Recent stress and the rs6295 G allele increase amygdala reactivity more than either alone.146 As well, DNA methylation of the C allele has been associated with reduced negative symptom response to antipsychotics in first-episode schizophrenia,147 which may account for an association with the C allele in 1 study.148 In agreement, several studies have shown that life stressors, including chronic pain or infection, interact with the rs6295 genotype for anxiety, depression and susceptibility to hospitalization for depression.51,81,146,149–151 However, in 1 study, although the G allele, childhood or later life stress were each associated with substance abuse, psychiatric hospitalization and suicide, there was no interaction between genotype and trauma in a highly stressed cohort.61 Similarly, other studies did not find an association between rs6295/stress and depression.152 An association with suicide or suicide attempt has been seen for rs6295 alone in some but not all studies,61,153,154 and in other studies the rs6295 genotype has been associated with suicide attempt or suicide only in people with previous trauma.155,156 The rs6295 HTR1A gene polymorphism is one of several SNPs that has been shown to interact with stress exposure in depression susceptibility,67 each study showing a stress-associated increase in depression frequency for risk alleles. However, as proposed for the 5-HTTLPR, this association may be strongest with persistent or recurrent depression rather than with single-episode depression. 157
Epigenetic mechanisms: clinical findings and animal models
Environmental stress can affect gene expression through epigenetic changes such as histone modification and DNA methylation.158,159 Histone acetylation mediates chromatin opening, typically inducing gene activation, while histone deacetylation closes chromatin to mediate gene repression. 160 The DNA methylation of promoters mediates recruitment of methyl-binding proteins to repress gene expression, while methylation of specific repressor sites can reduce gene activation or repression. For example, pups fostered by rats bred for enhanced maternal care show increased 5-HT activity, leading to reduced stress reactivity in adulthood.161 This persistent behavioural change involves reduced expression of hippocampal glucocorticoid receptors due to DNA methylation at a specific site in the gene.162 In humans, early-life abuse is associated with increased methylation of the analogous site in the brains of people who have died by suicide.163 The effects of 5-HT1A receptors on stress-induced modifications, and of chronic stress on 5-HT1A receptor gene transcription, histone acetylation and DNA methylation are presented in the sections that follow.
Stress and HTR1A epigenetic regulation: histone acetylation
The effects of chronic stress on epigenetic modifications of the HTR1A gene and depression-like behaviour have been examined in several studies. In addition, several lines of evidence implicate 5-HT1A receptors in modifying the histone acetylation of other genes following stress. The ability of 5-HT1A agonists to promote resilience to restraint stress was associated with increases in hippocampal histone-H3 acetylation and was mimicked by trichostatin-A, an inhibitor of histone deacetylase (HDAC).164 Similarly, 5-HT1A signalling in visual cortex reduces HDAC5 levels to trigger neural plasticity in the visual cortex of adult rats with monocular deprivation. 128 Interestingly, HDAC5 is increased in the PFC following chronic stress, and its inactivation has been implicated in antidepressant response to SSRI, tricyclic antidepressants and ketamine.165–167 Thus, 5-HT1A-induced signalling to histonemodifying enzymes through unexplored mechanisms may induce resilience to stress-induced depression.
The HTR1A gene itself is subject to regulation by histone acetylation. In non-neuronal cells that do not express 5-HT1A receptors, repression of HTR1A is HDAC-dependent. In contrast, in neuronal cells that do express the receptor, 5-HT1A receptor gene repression is at least partly HDAC-independent.168,169 In particular, the repressor Freud-1 assembles different BRG1 chromatin remodelling complexes in non-neuronal versus neuronal cells to mediate HDAC-dependent and -independent repression, respectively. Neuronal cells show de-repression of the HTR1A gene upon depletion of Freud-1 or BRG1, while non-neuronal cells are resistant,169,170 reflecting silencing of 5-HT1A receptor expression in the latter. This reversible regulation of 5-HT1A receptor expression in neurons may account for the dynamic regulation of 5-HT1A autoreceptors by Freud-1 in vivo.113 Although pharmacological inhibition of HDAC has antidepressant effects, HDAC subtype- or cell-specific inhibition would be needed to specifically target depression-related transcriptional processes.171
Stress and HTR1A epigenetic regulation: DNA methylation
In addition to altered histone acetylation, chronic stress is correlated with changes in DNA methylation and gene expression and increased risk of major depression or suicide. 159 Several clinical studies have shown alterations in human HTR1A gene methylation associated with mental illness and changes in 5-HT1A receptor expression. Increased DNA methylation of the human HTR1A promoter in leukocytes has been reported in bipolar depression and schizophrenia. 172 Hypomethylation of the HTR1A promoter in lymphocytes from lupus erythematosus patients correlates with increased 5-HT1A expression.173 Specific methylation of the rs6295 site has been negatively correlated with negative symptom treatment response in schizophrenia.147 In this regard, DEAF1 binding to its site is attenuated by DNA methylation, 174 suggesting that increased 5-HT1A autoreceptor expression may be underlying this treatment resistance. More recently, stress-linked hypomethylation in blood cells of a site (CpG668) in the HTR1A gene body was correlated with resistance to 8-week escitalopram treatment in treatment-naive depressed patients.175 Thus, methylation of the 5-HT1A promoter or repressor sites can affect 5-HT1A receptor expression and correlate with schizophrenia and response to atypical antipsychotics. However, the statistical power in many of these trials was not sufficient, and larger studies are needed.
The effect of chronic stress on 5-HT1A receptor promoter methylation and RNA expression has been examined in only one animal model to date. Methylation of 24 CpG sites on the mouse Htr1a promoter was quantified following unpredictable chronic mild stress (UCMS) for 9 weeks in male Balb/c mice.176 Stressed mice showed increased DNA methylation of a single site located within an Sp4 element of the Htr1a gene that correlated with increased 5-HT1A RNA levels in both raphe and PFC (Fig. 2). The DNA methylation of this site prevented Sp4-induced repression and could account for the stress-induced increase in 5-HT1A RNA. As shown by transcriptionally upregulating 5-HT1A autoreceptors in the raphe-specific Freud-1 conditional knockout mice,113 the stress-induced expression of 5-HT1A autoreceptors could mediate the acquisition of depressed behaviour in UCMS mice. Interestingly, in the Freud-1 knockout study, mice were individually housed, a stressor that could contribute to the increase in 5-HT1A autoreceptors and the SSRI-resistant behavioural phenotype. In contrast, chronic treatment of the UCMS mice with the tricyclic antidepressant imipramine reversed the depression-like phenotype, perhaps because imipramine targets additional (e.g., noradrenergic) mechanisms compared with SSRIs. Imipramine also reversed HTR1A methylation in both raphe and PFC, but the increase in 5-HT1A RNA was reduced only in the latter. Thus, the stress-induced site-specific DNA methylation appears to alter 5-HT1A receptor expression by inhibiting transcription factor (Sp4) binding, and is reversible upon chronic imipramine treatment.176 However, additional mechanisms maintained the stress-induced upregulation of 5-HT1A autoreceptors in imipramine-treated mice that could confer a persistent vulnerability to relapse. In this regard, antidepressant-naive depressed participants and remitted depressed participants (off medication) both had elevated 5-HT1A binding compared with controls,23,177 suggesting increased 5-HT1A expression as a trait or scar (residual characteristic) of persistent depression vulnerability.
Since the effects of DNA methylation on gene expression are mediated in part by methyl-binding proteins, such as MeCP2,178 the role of MeCP2 in the regulation of 5-HT1A receptor expression, 5-HT regulation and behaviour was examined. 103 In addition to methylation-sensitive mechanisms, MeCP2 binds to DEAF1 and enhances its repressor and enhancer activities at the human and mouse 5-HT1A genes (Fig. 2). In mice lacking MeCP2 in adult 5-HT neurons, 5-HT1A autoreceptors were upregulated, indicating that MeCP2 plays a key role to repress 5-HT1A autoreceptor expression. Interestingly, these mice showed a sex-dependent anxiety–depression phenotype, suggesting additional sex-dependent actions of MeCP2 in 5-HT neurons.103
Epigenetic regulation of the 5-HT1A receptor may be synergistic with genotype-dependent regulation. In rs6295 heterozygous healthy human fetal and adult PFC, 5-HT1A RNA was preferentially transcribed from the C allele,61 as expected from the enhancer effect of DEAF1 at the C allele in this tissue. 39 However, in depressed tissue, C- and G-derived RNAs were equal (Fig. 3), suggesting that DEAF1 or its regulation is attenuated in depression, potentially by DNA methylation. 61 This suggests that epigenetic processes regulated by chronic stress or antidepressant treatment can modify genotype-associated resilience to major depression. However, as a therapeutic strategy, inhibition of DNA methylation is not specific enough.158 Instead, transient or cell-specific inhibition of DNA methylation could be used to augment responses driven by antidepressants to enhance their effectiveness, although this remains to be tested.

Model of the effects of genotype on the regulation of 5-HT1A receptor expression by promoters and the 3′-untranslated region. Shown is a model of a neuron in the PFC of a healthy or depressed person, with the most abundant genotypes of the HTR1A gene shown in the cell nucleus. Transcription of the rs6295 G allele results in RNA containing the rs878567 T allele and reduced splicing, while transcription of the C allele results in enhanced splicing to stabilize the RNA and increase the translation of 5-HT1A receptors. In the PFC of healthy people, the C allele is favoured, while in the PFC of people with depression they are equal, reducing splicing and 5-HT1A expression.
Posttranscriptional regulation of HTR1A expression
MicroRNA mechanisms
As suggested, multiple mechanisms — including gene polymorphisms and epigenetic changes — can regulate promoter activity by altering transcription factor binding (Fig. 2). In addition, posttranscriptional mechanisms — including changes in RNA-binding proteins, such as splice factors, and microRNAs (miRNAs) that target the 3′-untranslated region (UTR) — can alter RNA stability to influence gene expression and neurodevelopment.179 Recently, regulation of RNA stability by miRNAs has been implicated in major depression. 180,181 For example, miR-16 was shown to inhibit 5-HTT expression in vitro in human, mouse and rat cell lines,182,183 and chronic fluoxetine treatment in mice induced miR-16 in raphe, correlating with downregulated 5-HTT levels in raphe. 183 In 5-HT neurons compared to non-5-HT neurons, miRNA microarray showed that several miRNAs are depleted, including miR-16 and miR-135.184 Importantly, miR-135a targets conserved elements to reduce both 5-HTT and 5-HT1A receptor RNA levels, and it was upregulated by acute or chronic SSRI or imipramine treatment. Forced over-expression of miR-135a in the 5-HT neurons of adult male mice reduced 5-HTT and 5-HT1A expression and reversed the anxiety and depression phenotypes following social defeat, while knockdown of miR-135a had the opposite effect on nonstressed mice.184 In these miR-135a mouse models, reductions of both 5-HTT and 5-HT1A autoreceptors could contribute to the increased 5-HT levels and release that correlated with behavioural improvement, as shown in studies modifying 5-HTT or 5-HT1A expression separately.105,113,185 Importantly, the potential roles for human miR-16 and miR-135a in depression were suggested by reductions in the raphe of brains of people who had died by suicide compared to healthy controls.184 Furthermore, miR-135a, but not miR-16, was reduced in the blood of depressed patients and was increased by cognitive behavioural therapy but not SSRIs. Several miRNAs are altered in the blood of depressed versus healthy people, and may serve as functional biomarkers for major depression or treatment response.181 Furthermore, direct targeting of 5-HTT or 5-HT1A using siRNAs may provide a novel and effective form of molecular therapy for major depression involving dysregulation of these proteins or the miRNAs that target them.186 However, an important caveat to targeting microRNAs is that, like transcription factors, they have multiple gene targets that could result in adverse outcomes; hence, cell-specific targeting would be important.
Alternative splicing
More recently, attention has focused on alternative splicing as another regulator of gene expression in major depression.187 Alternative splicing is particularly abundant in the developing human brain188 and is altered in major depression, resulting in altered RNA structure and new protein variants.187
Our recent studies, in addition to confirming regulation by miR-135a, showed that the HTR1A gene is subject to a novel alternative splicing event that removes the miR-135 site189 (Fig. 3). This splicing event is surprising, because the HTR1A gene was initially characterized as an “intronless” gene.190–192 However, in the human (but not rodent) HTR1A 3′-UTR, we discovered 2 very similar splice variants that are regulated in opposite ways by the brain-enriched splice factors nSR100 (enhances splicing) and PTBP1 (inhibits splicing; Fig 3).189 The spliced HTR1A RNA variants are extremely stable compared with the unspliced version that is degraded rapidly, consistent with destabilization induced by miR-135.184 The level of the unspliced form is lower in the PFC than in the hippocampus and raphe, but its expression increases in depression, which would lead to reduced 5-HT1A receptor levels as seen in postmortem PFC tissue from people with depression who died by suicide and in PET imaging studies of depressed people.193 Importantly, the levels of splice factor nSR100 were reduced in depressed PFC tissue and could account for the reduced level of 5-HT1A splicing.189 Thus, both splicing and miRNA regulation of the HTR1A gene converge to determine HTR1A RNA stability, and reductions in both splice factors (nSR100) and miRNAs (miR-135) that occur in major depression could synergistically alter 5-HT1A protein levels (Fig. 3).
In addition to the convergence of splicing and miRNA, genotype may also influence splicing. The HTR1A rs878567 SNP is located in the 3′-UTR of the 5-HT1A receptor close to the splice site, and is in strong linkage disequilibrium with the rs6295 promoter polymorphism.61 The rs878567 polymorphism has been associated with major depression,46,47 schizophrenia,194,195 psychosis196 and depression after childhood physical abuse.197 While many of these associations are similar to those for rs6295, the association with schizophrenia and psychosis was not as robust for rs6295. These differences are consistent with different roles for these SNPs in transcription compared to RNA processing of the 5-HT1A receptor gene. Our preliminary data suggest that the rs879567 C allele (linked to the rs6295 C allele) is associated with greater splicing and thus expression of the 5-HT1A RNA, synergistic with the greater DEAF1-induced HTR1A transcription in the PFC (Fig. 3). This illustrates the potential importance of genotype in determining the extent of RNA splicing and stability, and how these posttranscriptional processes may be synergistic (or antagonistic) with linked genotypes that affect transcriptional activity.
Recently, the largest genome-wide association study for major depression identified 2 SNPs with genome-wide significance located in the RBFOX1 gene, a regulator of gene splicing.198 RBFOX1 sites are generally located in introns 200 to 300 bp from the splice site, and RBFOX1 regulates splicing in the nervous system by combining with brain-specific splice factors.199 It is antagonized by PTBP1, which is reduced in the PFC in major depression.189 Interestingly, in the human HTR1A intron there is an RBFOX1 site ( TGCATG) located about 270 bp from the splice acceptor site, suggesting its role in 5-HT1A RNA splicing or regulation, in combination with nSR100 and PTBP1 in major depression. These converging lines of evidence highlight the potential importance of alterations in splice factor expression to modify alternative splicing and gene expression, resulting in susceptibility to major depression.
Conclusion
In summary, the 5-HT1A receptor gene illustrates the multilevel gene transcription and RNA regulatory mechanisms that determine protein expression levels. Alterations in these mechanisms combine to change protein expression and can be influenced by genotype (polymorphisms in both promoter, introns and UTR) and stress/environment (leading to epigenetic changes such as histone acetylation and DNA methylation). Furthermore, changes in the upstream regulators (transcription factors, methyl-binding proteins, splice factors, miRNAs) can have a broad influence on the expression of many genes in a cell, a subset of which seem to more strongly confer risk for mental illness and provide diagnostic or prognostic markers. For example, recent evidence suggests that in some cases blood miRNA (including miR-135) can be used as a marker for major depression and seems to mirror miRNA changes in the brain.181
Ultimately, by understanding and targeting or bypassing gene regulatory mechanisms elucidated in animal models, more effective personalized treatments for mental illness may emerge. For example, in depressed patients with the HTR1A rs6295 polymorphism and/or showing increased raphe 5-HT1A autoreceptors, as seen in depressed males and at-risk off-spring, 200,201 a strategy to block these receptors (e.g., using SSRI with pindolol or vortioxetine), or bypass presynaptic 5-HT by directly targeting postsynaptic 5-HT1A receptors or by targeting other monoamines (e.g., noradrenaline, using SNRIs, tricyclic antidepressants) may be more efficient than SSRI treatment alone.11 In the future, directly targeting the 5-HT1A autoreceptor using intranasal 5-HT1A siRNA conjugated to SSRI for raphe targeting could be used for knockdown 5-HT1A receptor RNA.186 One can envisage screening patients’ blood samples for depression-related miRNAs and then targeting those brain miRNAs using intranasal therapeutics.181 These strategies have been tested in animal models, and could be applied to human depression. The evidence from animal models supports a knowledge-based approach to use biomarkers such as genotype, miRNA, receptor or brain imaging, to interrogate the molecular, genetic and cellular mechanisms underlying major depression. With this understanding, personalized strategies to target the underlying causes could be applied for more rapid and effective treatment.
Acknowledgements
The authors’ research was supported in part by grants from the Canadian Institutes of Health Research to P. Albert.
Footnotes
Competing interests: P. Albert sits on the JPN editorial board. He was not involved in the decision-making on this manuscript. No other competing interests declared.
Contributors: All authors reviewed and interpreted the literature. P. Albert wrote the article, which all authors reviewed. All authors approved the final version to be published and can certify that no other individuals not listed as authors have made substantial contributions to the paper.
- Received November 1, 2018.
- Revision received December 10, 2018.
- Accepted December 21, 2018.
References

{kind=link}
{kind=link}
{kind=link}
Article tools





