

Abstract
Background In addition to motor disability, another characteristic feature of Parkinson disease is the early appearance of psychiatric symptoms, including apathy, depression, anxiety and cognitive deficits; treatments for these symptoms are limited by the development of adverse effects such as impulse-control disorders. In this context, we investigated the orphan G protein-coupled receptor 88 (GPR88) as a novel therapeutic target.
Methods We used lentiviral-mediated expression of specifically designed microRNA to knock down Gpr88 in a translational male rat model of early Parkinson disease obtained by dopamine loss in the dorsolateral striatum as a result of 6-hydroxydopamine lesions. We evaluated the impact of Gpr88 knockdown on the Parkinson disease model using behavioural, immunohistochemical and in situ hybridization studies.
Results Knockdown of Gpr88 in associative territories of the dorsal striatum efficiently reduced alterations in mood, motivation and cognition through modulation of the regulator of the G-protein signalling 4 and of the truncated splice variant of the FosB transcription factor. Knockdown of Gpr88 also reduced allostatic changes in striatal activity markers that may be related to patterns observed in patients and that provide support for an “overload” hypothesis for the etiology of the psychiatric symptoms of Parkinson disease.
Limitations Behavioural tests assessing specific cognitive and motivational parameters are needed to further characterize the effects of the lesion and of Gpr88 knockdown in early-stage and advanced Parkinson disease models, presenting more extensive dopamine loss. Additional studies focusing on the direct and indirect striatal output pathways are also required, because little is known about the signalling pathways regulated by GPR88 in different striatal cell types.
Conclusion GPR88 may constitute a highly relevant target for the treatment of the psychiatric symptoms of Parkinson disease.
Introduction
Parkinson disease is classically defined as a motor disorder related to the neurodegenerative loss of dopamine innervation of the striatum, but it also involves psychiatric symptoms that represent a major burden for patients’ quality of life.1 Many of these symptoms have been grouped as part of a “hypodopaminergic” syndrome, which includes apathy, depression, anxiety and cognitive impairment and affects up to 85% of patients with Parkinson disease.2,3 Apathy in particular, defined as a decrease in motivational drive, has emerged as a core symptom of Parkinson disease and is often associated with cognitive impairment.4
A remarkable feature of these symptoms is that, because of the progressive nature of the neurodegeneration, they frequently emerge years before the onset of motor impairments, which appear when striatal dopamine loss exceeds 60% to 70%.5,6 Thus, at the time of diagnosis, which is elicited by the appearance of motor symptoms, 37% of patients already have depression, 27% have apathy, 17% have anxiety and 20% to 40% have cognitive dysfunction,6 redefining Parkinson disease as a quintessential neuropsychiatric disorder.7
Dopaminergic medications can efficiently treat some of these aspects of Parkinson disease,3,4 but their effect is based on nonphysiologic pulsatile dopaminergic stimulation (similar to the effect of L-DOPA substitutive therapy for motor symptoms) and is thus hindered by the frequent development of “hyperdopaminergic” symptoms such as impulse-control disorders.2 Furthermore, existing treatments for cognitive impairment have variable efficacy and limiting adverse effects.8 There is thus an unmet medical need for new treatments to manage these symptoms.
In this context, orphan G protein-coupled receptor 88 (GPR88) is emerging as a particularly suitable target. The GPR88 gene has been associated with bipolar disorder and schizophrenia,9 as well as with learning deficits and chorea.10 It is expressed mainly in the corticostriatal synapse of striatal medium spiny neurons,11 where it exerts an inhibitory activity through Gi/o coupling.12,13 Indeed, Gpr88 knockout (KO) mice are hypersensitive to dopamine receptor type 2 agonists and display impaired affective, goal-directed, cognitive and sensorimotor behaviours.12,14,15 We have also reported that Gpr88 knockdown (Gpr88-KD) reduces behavioural alterations in a rat model of schizophrenia.16 As well, we have more recently shown that Gpr88-KD decreases locomotor alterations in a rat model of Parkinson disease motor symptoms. 17 Therefore, because GPR88 exerts inhibitory control over key neurotransmitter systems that are altered in Parkinson disease, we further hypothesized that inhibiting GPR88 in the striatum could have an effect on the psychiatric symptoms of Parkinson disease.
To evaluate GPR88 as a therapeutic target for the psychiatric symptoms of Parkinson disease, we first developed a translational model of early Parkinson disease in rats by reproducing the loss of dopamine that is observed in the first stages of the disease, specifically affecting sensorimotor territories of the striatum,18 using stereotaxic injections of the 6-hydroxydopamine (6-OHDA) toxin. We then implemented Gpr88-KD in the dorsolateral sensorimotor or dorsomedial associative areas of the striatum (the latter being associated with psychiatric symptoms in Parkinson disease19) and evaluated effects at the behavioural and molecular levels.
Methods
The study design is shown in Figure 1; details are provided in Appendix 1, available at jpn.ca/190171-a1.

Experimental design and timeline. The time sequence of the different experimental procedures and tests is outlined in the upper section. The experimental groups (number of rats in parentheses) and the specific aims of the study are presented in the lower section. 6-OHDA = 6-hydroxydopamine; DLS = dorsolateral striatum; DMS = dorsomedial striatum; miR = micro RNA; miR-Gpr88 = micro RNA against Gpr88; miR-neg = micro RNA negative.
Animals
Animal studies were authorized by an ethical committee (APAFIS reference #3669–2016011817516297) and conducted in the in-house specific pathogen–free animal facility (agreement reference B75–13–19). Male Sprague–Dawley rats weighing 300 g at the beginning of experimentation were housed in standard conditions and handled throughout the study in compliance with the European Union 2010 animal welfare act, and the 2010/63 French directive. Procedures were reported following the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.20
Stereotaxic injections of 6-OHDA
Intrastriatal stereotaxic injections were performed bilaterally with 4 μL of a solution of 6-OHDA (3 μg/μL, 12 μg total per hemisphere) in saline + 0.02% ascorbic acid (all chemicals from Sigma) or with a control solution (saline + 0.02% ascorbic acid only).
Lentivirus production
The lentiviral vectors coexpressing specifically designed microRNA (miR) and emerald green fluorescent protein were generated at the in-house iVector platform, using the BLOCK-iT Pol II miR RNAi expression vector kit (Invitrogen) and injected 2 weeks after the 6-OHDA lesions were introduced.
Behavioural tests
The rats were tested for general motor and exploratory behaviour using an actimeter, for anhedonia using the sucrose preference test, for social interaction and selective attention using the social novelty discrimination task, for sensorimotor gating using the prepulse inhibition test, and for depression-associated behaviour using the forced swim test.
Immunolabelling and in situ hybridization
After the end of the behavioural procedures (2 to 4 days later), the rats were anesthetized in an isoflurane chamber and decapitated. Their brains were rapidly removed and snap-frozen for 90 seconds in isopentane at −55°C (Carlo Erba Reagents). Coronal cryosections (12 μm) of the striatum were used to control for the presence and extent of emerald green fluorescent protein signal in the targeted areas. The slices were then post-fixed in 4% paraformaldehyde for 30 minutes at 4°C to perform immunolabelling and in situ hybridization experiments.
Statistical analysis
Data from the experiments were analyzed using Prism 6.0 (GraphPad Software Inc.) and implementing different tests as detailed in Appendix 1.
Results
6-OHDA stereotaxic injections induce dopaminergic denervation
The 6-OHDA injections in the dorsolateral striatum (DLS) reproduced the partial loss of dopaminergic projections to sensorimotor territories of the striatum (posterior putamen) observed in early Parkinson disease21 by inducing a 48% mean reduction of tyrosine hydroxylase immunoreactivity over at least 1 mm on the anterior–posterior axis (t16 = 9.28, p < 0.001; multiple t tests followed by Holm–Sidak corrections; Fig. 2A and B). The neigh-bouring associative dorsomedial striatum (DMS) and limbic ventral striatum (VS) were not significantly affected by the lesion (DMS t16 = 1.82, p = 0.09; VS t15 = 0.43, p = 0.67; Fig. 2B).

Stereotaxic injections induce a restricted loss of nigrostriatal dopaminergic projections and an efficient knockdown of Gpr88 expression in discrete striatal areas. (A) Representative images of 6-OHDA-lesioned striata. Coronal sections stained for TH by immunofluorescence (+1.2 to +0.2 mm anterior to the bregma), superimposed on the Paxinos and Watson rat brain atlas for anatomic reference.22 (B) Quantification of the extent of TH signal loss in the DLS, DMS and VS (nsham + miR-neg = 7–8; n6-OHDA + miR-neg = 10). (C) Schematic representation of the lentiviral construct used to transduce the targeted striatal regions. (D) Representative images of DLS or DMS transduced striata. The lentiviruses were injected at 2 coordinates anterior to the bregma: +1.2 and +0.2 mm. The photographs were then superimposed on the Paxinos and Watson rat brain atlas for anatomic reference.22 (E) ISH showing the suppression of Gpr88 expression induced by miR-Gpr88, but not miR-neg. (F) Quantification of the ISH signal for Gpr88 mRNA following miR-neg or miR-Gpr88 injections in the DLS or DMS (nsham + miR-neg = 12; n6-OHDA + miR-neg = 8–10; n6-OHDA + miR-Gpr88 = 8–10). The values in B and F were normalized to those of the control group (sham + miR-neg) and expressed as percent change. They are presented as mean ± standard error of the mean and were analyzed using a 2-way analysis of variance followed by Tukey multiple comparison tests. ****p < 0.0001. 6-OHDA = 6-hydroxydopamine; AP = anteroposterior; DLS = dorsolateral striatum; DMS = dorsomedial striatum; emGFP = emerald green fluorescent protein; ISH = in situ hybridization; LTR = long terminal repeat; LV = lentivirus; miR = micro RNA; miR-Gpr88 = micro RNA against Gpr88; miR-neg = micro RNA negative; NS = not significant; PGK Pol II = phosphoglycerate kinase polymerase II promoter; shRNA = short hairpin RNA; SIN/ΔU3 = self-inactivating deletion in the 3′ long terminal repeat; TH = tyrosine hydroxylase; VS = ventral striatum.
Lentivirus-mediated knockdown efficiently reduced Gpr88 expression
The injections at multiple sites in the DLS or DMS of lentiviral vectors containing a control miR (miR-neg) or an miR directed against Gpr88 (miR-Gpr88) resulted in the transduction of the corresponding regions without spreading to the adjacent areas (Fig. 2C and D). Two-way analysis of variance (ANOVA) showed significant effects of Gpr88-KD (F2,52 = 28, p < 0.001), the striatal region (F1,52 = 10, p = 0.002) and a Gpr88-KD × striatal region interaction (F2,52 = 72, p < 0.001) on Gpr88 expression. The region-specific effect of Gpr88-KD was confirmed using Tukey post hoc multiple comparison tests (p < 0.001 in the DLS and DMS; Fig. 2E and F).
Partial dopaminergic depletion of the DLS reproduced psychiatric symptoms without inducing motor deficits
Motor behaviour was assessed at 2 and 4 weeks after 6-OHDA injections by measuring horizontal movements in the actimeter. This parameter was unaffected by dopamine loss at both time points (Fig. 3A; Appendix 1, Fig. S2A), as expected with a restricted lesion.23 However, although stereotyped behaviour in the actimeter test was also preserved at both time points, 2-way ANOVA revealed a significant interaction of treatment × time on the number of rearings (F2,44 = 4.66, p = 0.015; Fig. 3A); these were significantly reduced by the lesion at the 15-minute time point (Sidak multiple comparison test, p = 0.013), indicating a decrease in novelty exploration behaviour, previously characterized in similar models of partial dopamine loss in the DLS.24,25

Dopamine loss in the dorsolateral striatum reproduces psychiatric symptoms of Parkinson disease. (A) Horizontal, stereotyped and rearing behaviours measured during 3 consecutive 5-minute segments with the actimeter test 4 weeks after the 6-OHDA lesion (nsham + miR-neg = 14; n6-OHDA + miR-neg = 10). (B) Effects of the 6-OHDA lesion on the startle amplitude in response to a loud stimulus (110 dB) and on percent of prepulse inhibition at all tested prepulse intensities (70, 75 and 80 dB; nsham + miR-neg = 14; n6-OHDA + miR-neg = 7–10). (C) The 6-OHDA lesions did not alter the percent preference for the sucrose solution (nsham + miR-neg = 6; n6-OHDA + miR-neg = 7). (D) The 6-OHDA lesions did induce depressive-like behaviour in the forced swim test, as suggested by decreased latency (in seconds) and increased immobility count (nsham + miR-neg = 16; n6-OHDA + miR-neg = 10). (E) The 6-OHDA lesions also decreased P1 investigation time (in seconds) in the social novelty discrimination task, abolished preference for the novel juvenile and decreased the discrimination ratio (nsham + miR-neg = 16; n6-OHDA + miR-neg = 10). Data are presented as mean ± standard error of the mean. When comparing 2 groups, we performed 2-tailed Welch t tests. In cases of interactions with additional factors, we used 2-way analyses of variance, followed by a Sidak multiple comparison test (actimeter data, percent prepulse inhibition and P2 investigation time). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. 6-OHDA = 6-hydroxydopamine; a.u. = arbitrary units; miR = micro RNA; miR-neg = micro RNA negative; P1 = first presentation; P2 = second presentation.
Sensorimotor gating is altered in many basal ganglia disorders, including Parkinson disease, and has been linked to cognitive dysfunction in patients with Parkinson disease.26 We assessed this parameter using the prepulse inhibition test. Although the partial lesion of the DLS increased the startle reaction in response to a loud auditory stimulus (110 dB; t10.21 = 2.396, p = 0.037, Welch t test; Fig. 3B, left panel), the level of sensorimotor gating (% prepulse inhibition) was preserved at all levels of prepulse intensity used (Fig. 3B, right panel), suggesting the presence of efficient compensatory mechanisms.
Affective parameters relating to anhedonia and depression (both frequently reported in Parkinson disease)3 were assessed using the sucrose preference and the forced swim tests, respectively. Sucrose preference and general consummatory behaviour, measured by daily food and water intake, were not affected by the 6-OHDA lesion (Fig. 3C; Appendix 1, Fig. S2B). However, the lesion did decrease the latency to immobility and increased immobility count in the forced swim test (latency t21.34 = 3.94, p < 0.001; immobility count t22.85 = 4.36, p < 0.001; Welch t tests; Fig. 3D), representative of a despair-like behaviour associated with depression.27
Finally, we used the social novelty discrimination task to evaluate deficits in motivation and selective attention (a core feature of Parkinson disease executive dysfunction).8 The loss of dopamine in the DLS decreased social interaction time during the first presentation phase (P1; t22.72 = 2.95, p = 0.007, Welch t test; Fig. 3E), an ecological indicator of apathy.28 Further analyses then revealed a significant treatment × novelty status interaction in the discrimination phase of the test (P2; 2-way ANOVA: F1,48 = 8.995, p = 0.004); the lesioned animals failed to preferentially direct their attention to the novel juvenile, which resulted in a decreased discrimination ratio (t22.51 = 4.29, p < 0.001, Welch t test; Fig. 3E), an effect that was not due to a reduced level of interaction during the discrimination task (Appendix 1, Figure S2C).
Gpr88-KD in the DMS reversed behavioural deficits
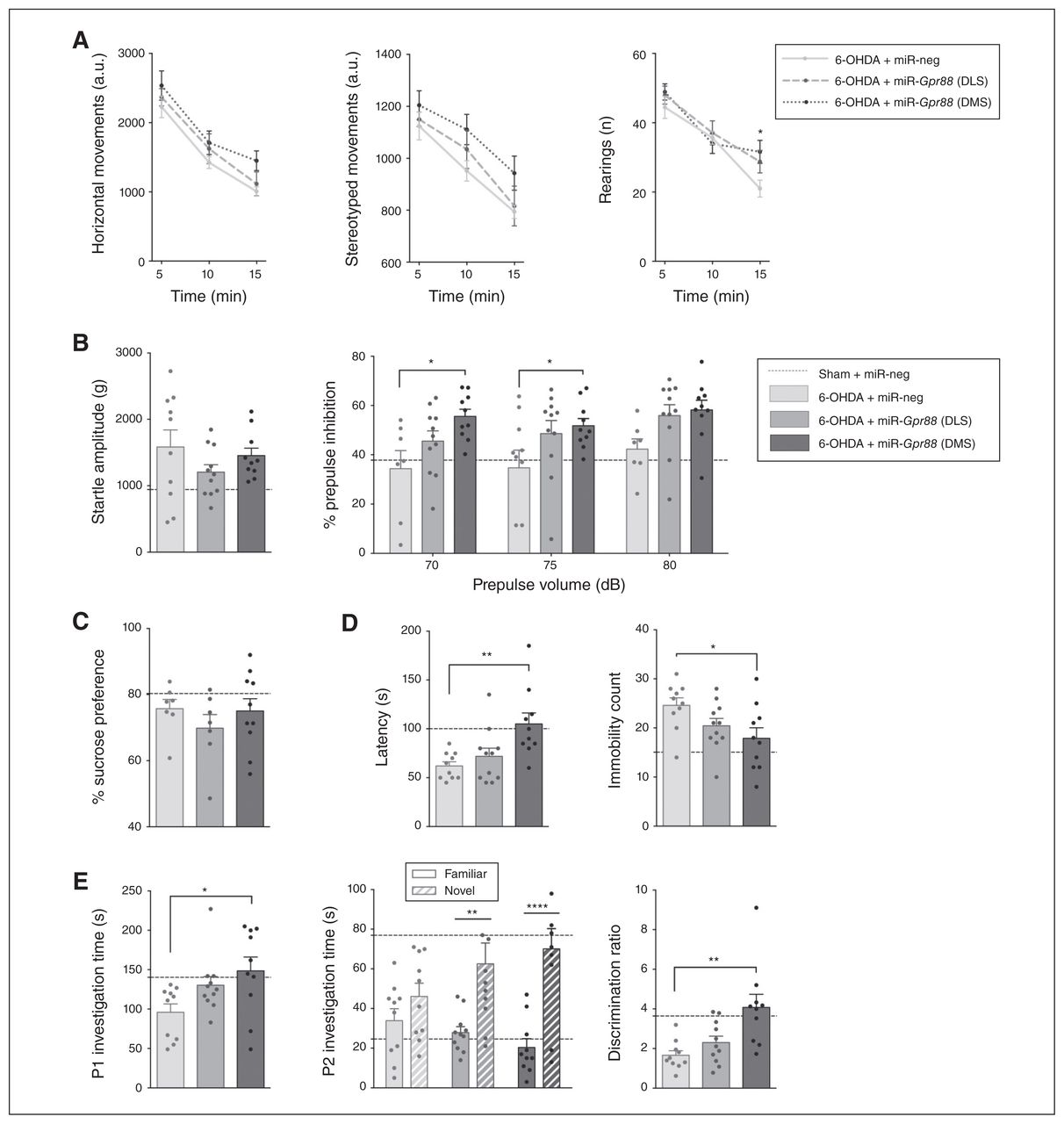
Although 2-way ANOVA revealed no significant main effects of treatment or treatment × time interaction on the motor parameters measured by the actimeter, Gpr88-KD in the DMS reversed the deficit in rearing behaviour at the 15-minute time point (p = 0.020, Dunnett post hoc multiple comparison test; Fig. 4A). It also increased sensorimotor gating without affecting startle amplitude (2-way ANOVA, effect of treatment on % prepulse inhibition, F2,78 = 9,79, p < 0.001; Dunnett post hoc test miR-Gpr88-DMS v. miR-neg, 70 dB p = 0.010 and 75 dB p = 0.025; Fig. 4B).
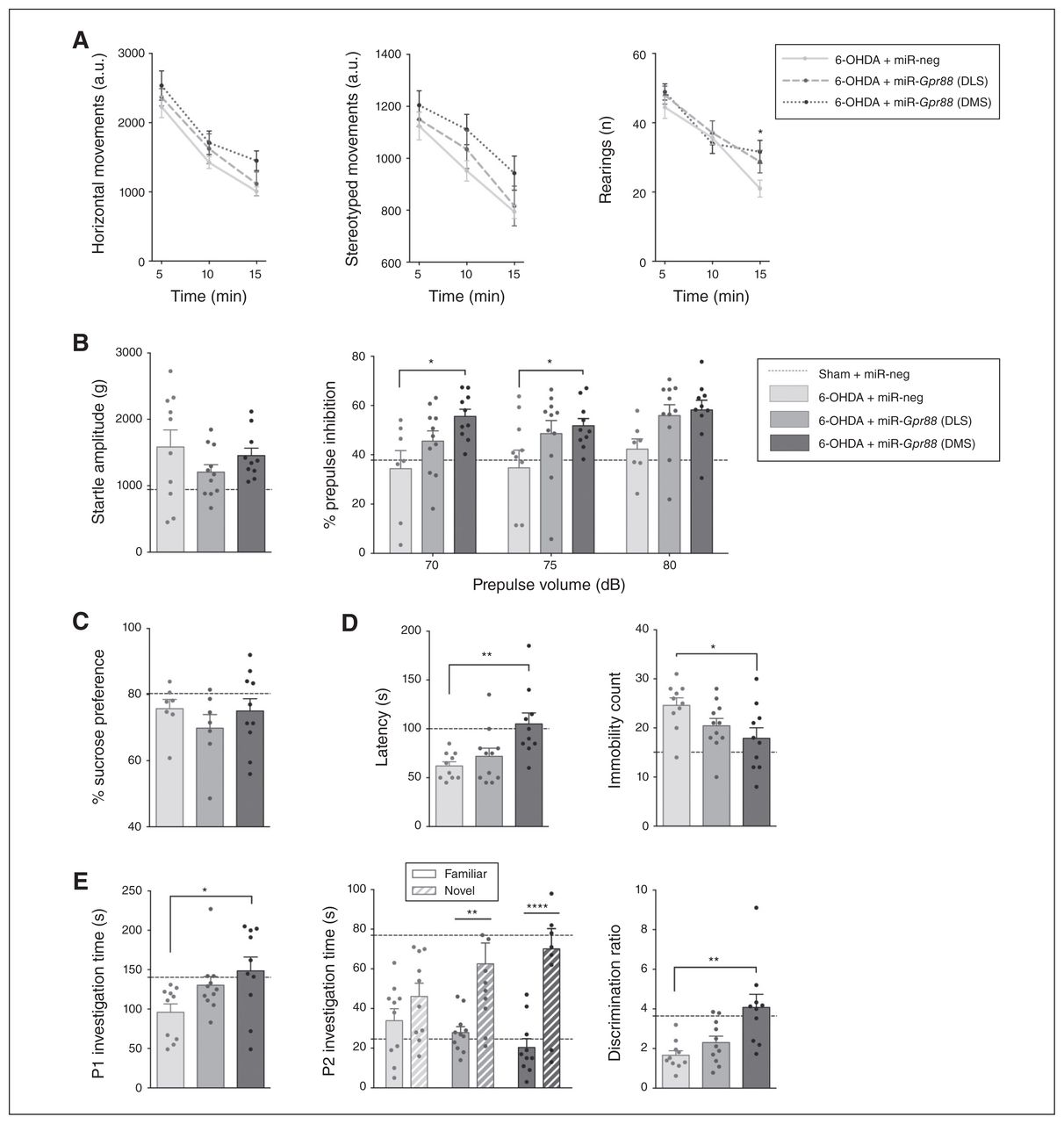
Gpr88 knockdown in the dorsomedial striatum reverses behavioural deficits. Effects of Gpr88 knockdown on the following: (A) horizontal, stereotyped and rearing behaviours in the actimeter test; (B) startle amplitude and percent of prepulse inhibition; (C) sucrose preference; (D) latency and immobility count in the forced swim test; (E) social interaction and novelty preference; (n6-OHDA + miR-neg = 10; n6-OHDA + miR-Gpr88-DLS = 9–11; n6-OHDA + miR-Gpr88-DMS = 8–10, except in the sucrose preference test, where n = 7–10 throughout the groups). Data are presented as mean ± standard error of the mean. For reference, a dashed horizontal line indicates the values from the control group (Sham + miR-neg) that were presented in Figure 3. When we compared the 3 groups for 6-OHDA, we performed 1-way analyses of variance followed by a Dunnett multiple comparisons test. In cases of interactions with additional factors, we performed 2-way analyses of variance, followed by a Dunnett multiple comparisons test (actimeter data, % prepulse inhibition and P2 investigation time). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. 6-OHDA = 6-hydroxydopamine; a.u. = arbitrary units; DLS = dorsolateral striatum; DMS = dorsomedial striatum; miR = micro RNA; miR-neg = micro RNA negative; miR-Gpr88 = micro RNA against Gpr88; P1 = first presentation; P2 = second presentation.
Furthermore, whereas Gpr88-KD in either area had no effect on sucrose preference (Fig. 4C), inactivation in the DMS had an antidepressant-like effect in the forced swim test: it increased latency (effect of treatment F2,28 = 6.883, p = 0.004; Dunnett post hoc test miR-Gpr88-DMS v. miR-neg p = 0.003) and reduced immobility count (effect of treatment F2,28 = 3.736, p = 0.037; Dunnett post hoc test miR-Gpr88-DMS v. miR-neg p = 0.021; Fig. 4D). This reduction was specifically mediated by an increase in swimming behaviour (effect of treatment F2,28 = 3.524, p = 0.043; Dunnett post hoc test miRGpr88-DMS v. miR-neg p = 0.028; Appendix 1, Fig. S3A). We observed no effect on consummatory behaviour (Appendix 1, Fig. S3B).
In the DMS, Gpr88-KD also had a promotivational effect, as it increased the duration of social interaction (effect of treatment F2,28 = 3.938, p = 0.031; Dunnett post hoc test miR-Gpr88-DMS v. miR-neg p = 0.019; Fig. 4E). Finally, Gpr88-KD increased preferential interaction with the novel juvenile in the social novelty discrimination task (2-way ANOVA, treatment × novelty status interaction F2,56 = 3.162, p = 0.05) in both the DLS and DMS (p= 0.004 and p < 0.001, respectively; Sidak multiple comparison test; Fig. 4E). However, only the effect in the DMS was large enough to restore the discrimination ratio (effect of treatment F2,28 = 7.998, p = 0.002; Dunnett post hoc test miR-Gpr88-DMS v. miR-neg p = 0.001; Fig. 4E). However, this proattentional effect was not associated with an increase in total interaction time during the discrimination task (Appendix 1, Fig. S3C). It is worth noting that although significant effects of Gpr88-KD were observed mainly when targeting the DMS, inactivation in the DLS often also tended to reduce behavioural alterations.
Dopamine loss in the DLS induces molecular alterations in the adjacent DMS
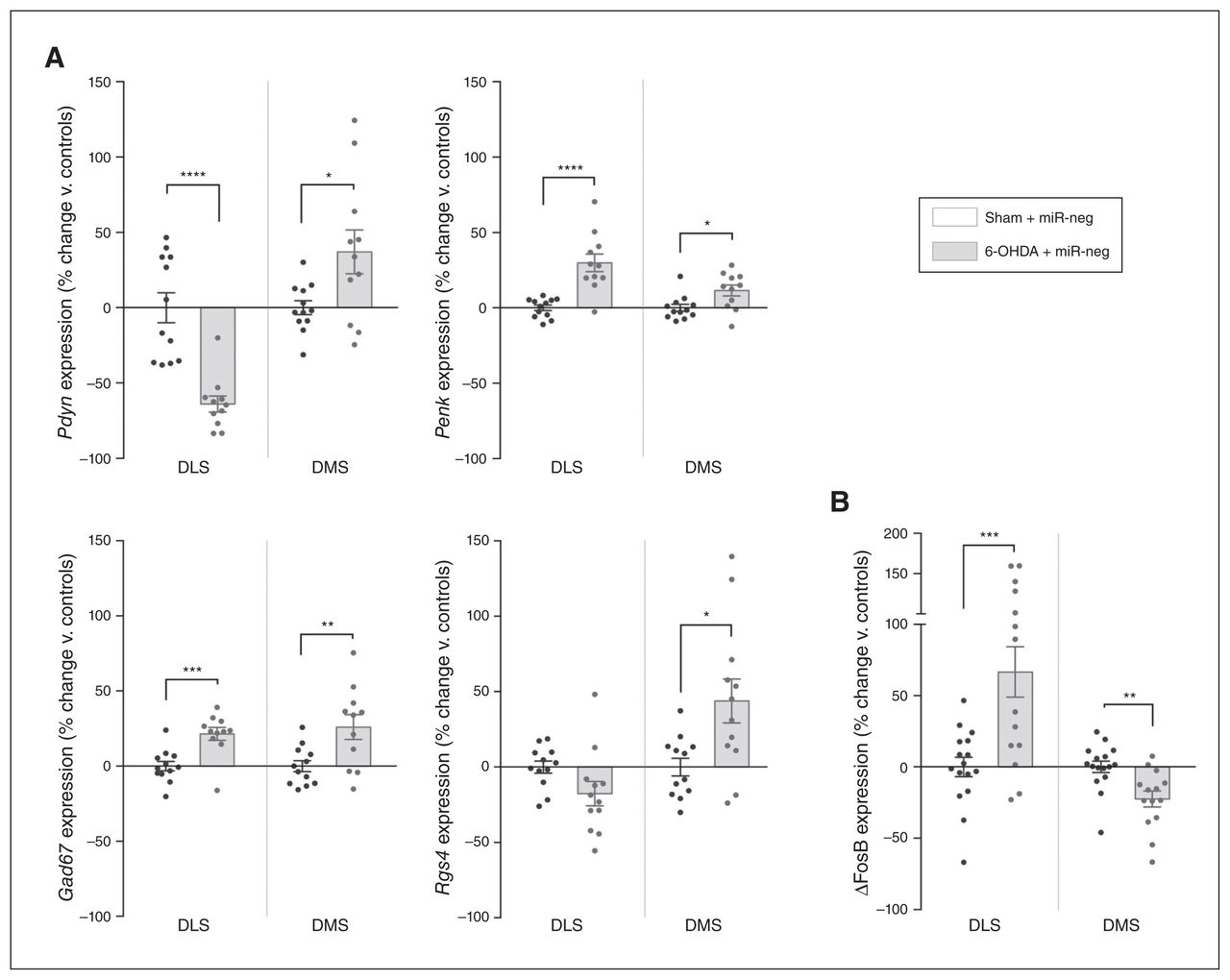
The dopamine striatal loss in Parkinson disease or after a 6-OHDA lesion dysregulates the balanced activity of the 2 types of projecting medium spiny neurons — the direct stimulatory striatonigral (dMSN) and the indirect inhibitory striatopallidal (iMSN) pathways — inducing the symptoms of Parkinson disease.29 Alterations in these pathways are associated with the downregulation of prodynorphin (Pdyn) in the dMSN and the upregulation of proenkephalin (Penk) in the iMSN, as well as a net increase in striatal glutamic acid decarboxylase 67 (Gad67) expression.30 Therefore, we evaluated the expression levels of Pdyn and Penk, which reflect dMSN and iMSN activity, respectively,31 and Gad67, which is considered a proxy for global levels of transmission of γ-aminobutyric acid in the striatum.32 Strikingly, although the loss of dopamine was strictly limited to the DLS, we also observed significant alterations in the expression of these markers in the adjacent, intact DMS. Indeed, although the lesion decreased expression levels of Pdyn and increased those of Penk in the DLS as expected (Pdyn t21 = 5.551, p < 0.001; Penk t21 = 5.035, p < 0.001; multiple t tests followed by Holm–Sidak corrections, here and throughout the next sections; Fig. 5A), expression of both markers was upregulated in the unlesioned DMS (Pdyn t21 = 2.522, p = 0.020; Penk t21 = 2.709, p = 0.013; Fig. 5A), suggesting a local hyperactivity of dMSN and iMSN. Furthermore, Gad67 expression was also significantly upregulated throughout the dorsal striatum (DLS t21 = 4.095, p < 0.001; DMS t21 = 2.967, p = 0.007; Fig. 5A).
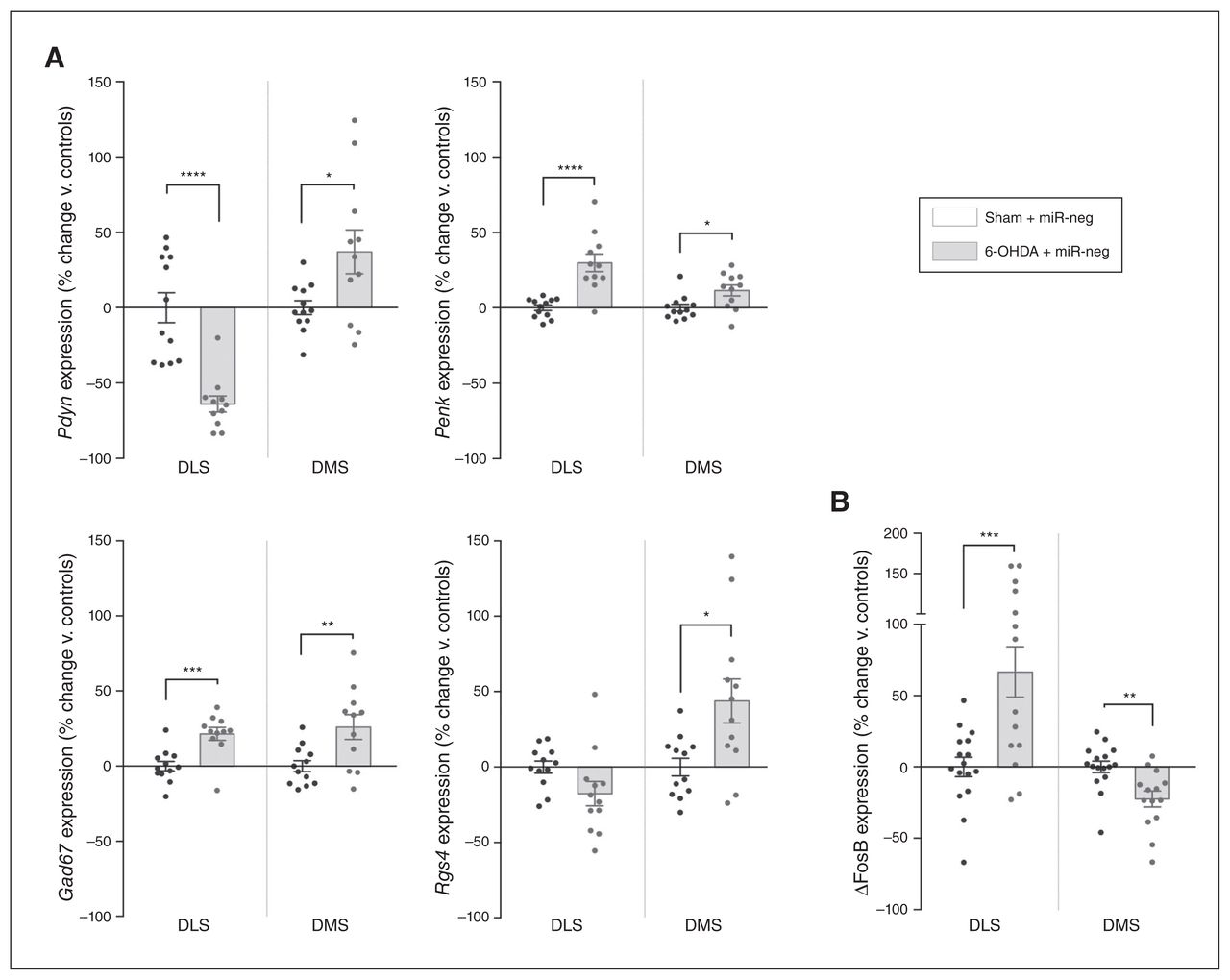
Molecular changes induced by dopamine loss in the DLS also affect the adjacent DMS. (A) Expression of Pdyn, Penk, Gad67 and Rgs4 (n = 12 in each condition). (B) Expression of ΔFosB in the DLS and DMS of 6-OHDA lesioned rats (nsham + miR-neg = 16; n6-OHDA + miR-neg = 14). Values were normalized to those of the control group (Sham + miR-neg) and expressed as percent change. Data are presented as mean ± standard error of the mean, and were compared using multiple t tests with Holm–Sidak corrections. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. ΔFosB = truncated splice variant of FosB; 6-OHDA = 6-hydroxydopamine; DLS = dorsolateral striatum; DMS = dorsolateral striatum; miR = micro RNA; miR-neg = micro RNA negative.
The regulator of G protein signalling 4 (Rgs4) is highly expressed in the brain and acts as a negative regulator of synaptic signalling.33 In 6-OHDA models of Parkinson disease, inhibition of Rgs4 has antiparkinsonian effects: it reduces behavioural alterations by restoring long-term depression in the iMSN while reducing acetylcholine release from cholinergic interneurons.34,35 Moreover, the increase in the excitability of medium spiny neurons associated with Gpr88-KO is mediated by the downregulation of Rgs4.12 Thus, we used Rgs4 expression to further investigate the effects of the lesion and Gpr88-KD on intracellular signalling. The 6-OHDA lesion induced a trend toward a decrease in the expression of Rgs4 in the DLS (t22 = 1.965, p = 0.06) and significantly increased expression in the DMS (t22 = 2.794, p = 0.011; Fig. 5A). However, Gpr88 expression remained unaffected by the loss of dopamine in either striatal area (Appendix 1, Fig. S4).
We then used the truncated splice variant of FosB (ΔFosB), a transcription factor involved in neuropsychiatric disorders including Parkinson disease,36 as a final molecular readout, because its unique accumulation profile confers a long-lasting effect on the regulation of striatal gene networks. 37 We found that ΔFosB expression was locally increased by the loss of dopamine in the DLS (t28 = 3.713, p = 0.002), but it was also significantly decreased in the unlesioned DMS (t28 = 3.338, p = 0.002; Fig. 5B).
Inactivation of Gpr88 in the DMS reverses alterations in Rgs4 and ΔFosB expression
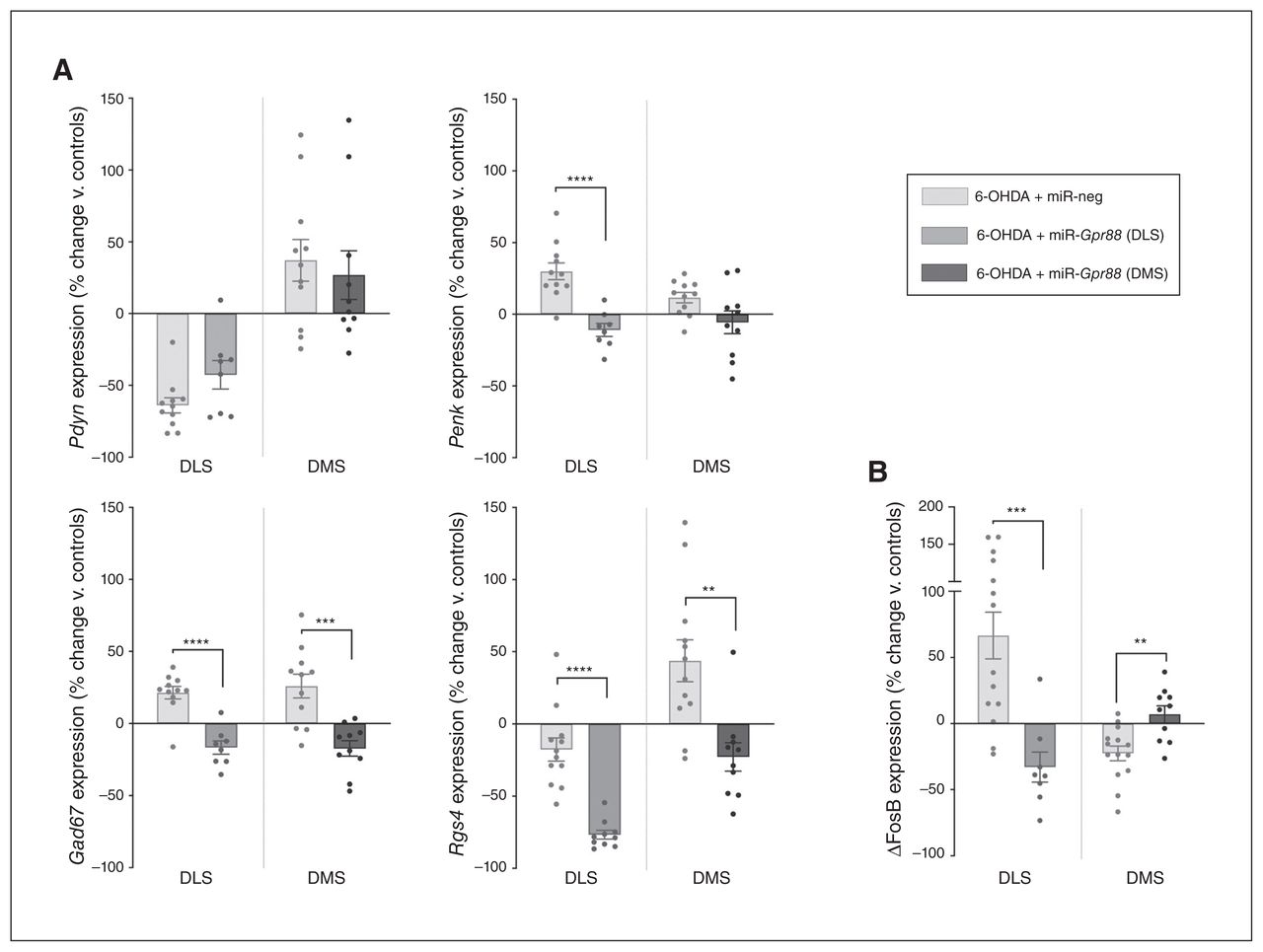
Although Gpr88-KD had no significant effect on Pdyn expression, it significantly reduced the expression level of Penk in the DLS (t17 = 5.163, p < 0.001; Fig. 6A), suggesting a reduction of iMSN hyperactivity. Also, Gpr88-KD strongly reduced the expression of Gad67 and Rgs4 in the DLS and DMS (DLS t17 = 5.956, p < 0.001 for Gad67 and t20 = 6.404, p < 0.001 for Rgs4; DMS t19 = 4.319, p < 0.001 for Gad67 and t20 = 3.633, p = 0.002 for Rgs4; Fig. 6A). Interestingly, Gpr88-KD had an opposing effect on ΔFosB expression in the DLS and DMS, in both cases reversing the 6-OHDA-induced alterations (t20 = 3.982, p < 0.001 and t22 = 3.452, p = 0.002, respectively; Fig. 6B).

Gpr88 knockdown modulates markers of neuronal activity, signalling and transcription. (A) Expression of Pdyn, Penk, Gad67 and Rgs4. (B) Expression of ΔFosB in the DLS and DMS after Gpr88 knockdown (n6-OHDA + miR-neg = 12–14, n6-OHDA + miR-Gpr88 = 8–10. Values were normalized to those of the control group (Sham + miR-neg; shown in Fig. 5) and expressed as percent change. Data are presented as mean ± standard error of the mean, and were compared using multiple t tests with Holm–Sidak corrections. **p < 0.01, ***p < 0.001, ****p < 0.0001. ΔFosB = truncated splice variant of FosB; 6-OHDA = 6-hydroxydopamine; DLS = dorsolateral striatum; DMS = dorsolateral striatum; miR = micro RNA; miR-neg = micro RNA negative; miR-Gpr88 = micro RNA against Gpr88.
Discussion
Although the acute dopamine loss in the 6-OHDA lesion model does not correspond to the progressive course of Parkinson disease, the partial bilateral striatal dopamine denervation most closely mimics the disease’s early stage.38 Accordingly, we established that the loss of dopamine restricted to the DLS was sufficient — without modifying motor behaviour — to reproduce alterations relating to depression, apathy and attentional deficits, 3 of the most prevalent psychiatric symptoms at the time of Parkinson disease diagnosis,39 and that Gpr88-KD can reduce several of these deficits.
Indeed, dopamine loss increased the immobility count in the forced swim test, an indicator of behavioural despair linked to depression.27 It also induced a decrease in social interaction and rearing behaviour related to novelty exploration, 24 2 parameters that have been proposed as ecological and translational measures of apathy.28 These results were also consistent with observations that the behavioural and social aspects of apathy are the most affected in Parkinson disease, and that they are specifically associated with depression. 40 Furthermore, apathy in Parkinson disease is frequently associated with cognitive impairment and dementia. 4 In this regard, we also observed that the lesioned animals performed poorly in the social novelty discrimination task, suggesting an impairment of selective attention41 — a core component of the dysexecutive syndrome experienced by patients with Parkinson disease.8
However, we did not reproduce the deficits relating to anhedonia (sucrose preference) and sensorimotor gating (prepulse inhibition) that had been previously reported in other 6-OHDA models of Parkinson disease.42,43 Anhedonia in Parkinson disease has been associated with alterations in the mesolimbic dopamine pathway,44 and prepulse inhibition alterations require extensive dopamine loss,42 suggesting that the lack of prepulse inhibition and sucrose preference alterations in our model were due to the restricted nature of the lesion.
Importantly, although the inactivation of Gpr88 in the lesioned DLS tended to have an effect on some of the altered behaviours, only the knockdown in the intact associative DMS reversed all of the behavioural deficits induced by dopamine loss, suggesting an effect that could be related to functional cross-talk between these 2 regions.45 In the DMS, Gpr88-KD also increased sensorimotor gating in the prepulse inhibition test, indicating an increased efficiency of preattentive information processing, which could underlie the pro-attentional effect in the social novelty discrimination task. Interestingly, computational models of Parkinson disease psychosis have linked the emergence of hallucinations to deficits in such gating and attentional processes,46 suggesting that Gpr88-KD may constitute a relevant target for evaluating this symptom in animal models.
The restricted loss of dopamine inputs to the sensorimotor territories of the striatum also altered the expression of molecular markers of neuronal activity, signalling and transcription beyond the lesioned area, significantly affecting associative territories. For instance, although the lesion locally induced a decrease in Pdyn and an increase in Penk and Gad67 expression levels (reproducing the imbalance in neuronal activity that has been characterized in Parkinson disease models31,32), their expression was significantly elevated in the DMS, suggesting a compensatory overactivity of neurons in response to dopamine depletion in the DLS.
A previous study had reported that 6-OHDA lesions induced a decrease in Gpr88 mRNA and protein expression after 1 and 4 weeks,11 but we did not observe such an effect at 6 weeks, suggesting a time-dependent recovery of Gpr88 expression following dopamine loss. The Gpr88-KD in the lesioned DLS was associated with a significant decrease in the expression of Penk and Gad67, suggesting that Gpr88-KD preferentially affects iMSN, which is consistent with the receptor’s relative enrichment in this neuronal type11 and the hypersensitivity to dopamine receptor type 2 agonists that has been consistently reported in Gpr88-KO mice.12,14 Because GPR88 is also known to modulate enkephalin delta opioid receptor signalling,15 the reduction in Penk expression observed after Gpr88-KD may have resulted partly from a potentiation of this receptor’s effects. In the DMS, Gpr88-KD reduced Gad67 overexpression without significantly affecting Penk or Pdyn, suggesting a potential decrease in γ-aminobutyric acid interneuron activity,32 in which GPR88 expression has been also reported.11
A GTPase accelerating protein, RGS4 negatively regulates the signalling of Gi/o- and Gq-associated receptors such as dopamine receptor types 2 and 3,47 metabotropic glutamate receptor type 548 and muscarinic acetylcholine receptor M4.35 Previous studies have reported decreases in striatal RGS4 mRNA in people with Parkinson disease49 and in 6-OHDAlesioned rodents.50 Although Rgs4 expression was slightly decreased in the DLS (p = 0.06), it was also strikingly elevated in the DMS. However, this overexpression was completely reversed by Gpr88-KD, an effect that was in line with findings from knockout mice.12,15 Interestingly, in models of Parkinson disease, inhibition or loss of Rgs4 has been shown to have potent antiparkinsonian effects,51 notably by restoring long-term depression in iMSN.34 However, translation to the clinic of these findings has been impeded by the high risk of adverse effects due to the ubiquitous presence of RGS4. Therefore, targeting GPR88 may constitute a unique strategy for bypassing these limitations, by inhibiting RGS4 with high spatial relevance, because GPR88 is exclusively expressed in the central nervous system and is mostly enriched in the striatum, where its expression levels match the typical pattern of dopamine loss.52
The 6-OHDA lesion and Gpr88-KD also had contrasting effects on the expression of transcription factor ΔFosB. Although ΔFosB accumulation in Parkinson disease has been linked to the effects of L-DOPA treatment,36 we also observed an increase in the DLS of the lesioned rats. This unexpected effect could have been due to stimulation of hypersensitive D1 receptors in the DLS, resulting from a “volume transmission” effect from the unlesioned striatum.53 In the DMS, however, we found a significant decrease in ΔFosB expression, which was reversed by Gpr88-KD. Interestingly, the anti-depressant effect of Gpr88-KD observed in the forced swim test may have been due to its modulation of ΔFosB, because fluoxetine is also known to increase the expression of this transcription factor in the DMS.54
The finding that the knockdown of Gpr88 in the associative striatum reduced behavioural deficits and normalized the expression of Rgs4 and ΔFosB must be integrated with our other recent work showing that Gpr88-KD in the dorsal striatum has an antiparkinsonian-like effect in a model of the motor symptoms of Parkinson disease by regulating the expression of Pdyn, Penk and ΔFosB.17 Thus, although the models we used are not easily comparable, taken together our results indicate that GPR88 may represent an unique target for treating both the psychiatric and the motor symptoms of Parkinson disease and avoiding some of the short-comings of current therapies.
Moreover, this study’s findings may also provide insights into the etiology of the psychiatric symptoms of Parkinson disease. Indeed, although psychiatric symptoms have been linked to alterations in multiple neurotransmitter systems,19 clustering studies of de novo patients suggest that the loss of striatal dopamine in particular may play an important role in their emergence.55 This idea is further supported by studies in rodent models, which have consistently shown that inducing extensive dopamine loss throughout the striatum could induce representative behavioural deficits.43 Nevertheless, these models have not necessarily accounted for the frequent premotor appearance of these symptoms, when dopamine loss is limited mainly to the sensorimotor areas of the striatum. 18 In this regard, our results show that such a restricted loss of dopamine is in itself sufficient to reproduce these behavioural alterations in an animal model.
However, these psychiatric aspects of Parkinson disease have typically been associated with dopamine dysfunction in associative networks (DMS),19 which are relatively preserved in the early stages of the disease, as well as in our model. Nevertheless, the overexpression of neuronal activity markers that we observed in the DMS suggests possible interactions between striatal areas. Interestingly, some functional MRI studies in people with Parkinson disease have also found that although functional connectivity decreased in the dopamine-deprived sensorimotor networks of the basal ganglia, it was increased in associative networks.56,57 These increases are thought to reflect early Parkinson disease allostatic mechanisms, by which associative networks expand or strengthen their connectivity to compensate for the loss of dopamine in sensorimotor territories. Although the underlying mechanisms are currently unknown, recently characterized striatonigrostriatal cellular pathways may provide a system for such lateral transfer of information.58 However, because of the resulting overlap of networks within limited striatal computational resources, it has been suggested that this compensatory mechanism could lead to a “neural bottle-neck” or “overload” of associative networks, and an impairment in related behaviours.21,57 This hypothesis could explain the emergence of psychiatric deficits relating to cognition and motivation during the earliest stages of Parkinson disease and in the striatal 6-OHDA focalized-lesion model of Parkinson disease, before the loss of dopamine affects associative networks.
Although such imbalances in DMS/DLS dynamics are emerging as key players in the pathophysiology of basal ganglia disorders such as addiction59 and obsessive–compulsive disorder,60 this study is, to our knowledge, the first to enquire into this concept in a rodent model of Parkinson disease.
Limitations
Several factors need to be investigated further. For instance, behavioural tests assessing specific cognitive and motivational parameters are needed to better characterize the effects of the lesion and of Gpr88-KD. Because the model reproduced aspects of early-stage Parkinson disease, it will also be necessary to assess the effects of the Gpr88-KD in models of advanced Parkinson disease, presenting more extensive dopamine loss and alterations in other neurotransmitter systems. Furthermore, although molecular experiments have provided some clues about the mechanisms involved, additional in-depth studies are required, because little is known about the signalling pathways regulated by GPR88 in different striatal cell types (dMSN/iMSN/interneurons).
Conclusion
Although the restricted expression of GPR88 makes it an ideal therapeutic target, no antagonist has so far been developed for this orphan receptor. It is thus crucial to develop lead identification efforts. Nevertheless, gene-therapy approaches have shown successful applications in Parkinson disease.61 Based on our findings, GPR88 gene therapy could constitute a highly relevant non-dopaminergic strategy for treating the psychiatric and the motor symptoms of Parkinson disease through striatal-level modulation of allostatic basal ganglia function.
Acknowledgments
The lentiviral vectors were produced by the in-house iVector facility. In vivo experiments were carried out in the PHENOPARC core facility, and histological procedures were performed at the HISTOMICS platform, both also situated in the ICM. The authors thank Annick Prigent for her time and advice related to the histological processing of the samples.
Footnotes
Competing interests: None declared.
Contributors: B. Galet, P. Ravassard and R. Meloni designed the study. B. Galet, M. Ingallinesi, J. Pegon, A. Do Thi, N. Faucon Biguet and R. Meloni acquired the data, which B. Galet, A. Do Thi, N. Faucon Biguet and R. Meloni analyzed. B. Galet and R. Meloni wrote the article, which all authors reviewed. All authors approved the final version to be published and can certify that no other individuals not listed as authors have made substantial contributions to the paper.
Funding: The research leading to these results has received funding from the program Investissements d’avenir ANR-10-IAIHU-06. B. Galet was supported by a scholarship from the French Ministry of Research, awarded by the Brain-Cognition-Behavior Doctoral School of the Sorbonne University. The PHENOPARC Core is supported by 2 Investissements d’avenir grants (ANR-10-IAIHU-06 and ANR-11-INBS-0011-NeurATRIS) and the Fondation pour la Recherche Médicale. The funding sources had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
- Received October 17, 2019.
- Revision received February 3, 2020.
- Accepted March 3, 2020.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is non-commercial (i.e. research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.