

Abstract
Background Late-life depression is often associated with non-response or relapse following conventional antidepressant treatment. The pathophysiology of late-life depression likely involves a complex interplay between aging and depression, and may include abnormalities in cortical inhibition and plasticity. However, the extent to which these cortical processes are modifiable by antidepressant pharmacotherapy is unknown.
Methods Sixty-eight patients with late-life depression received 12 weeks of treatment with open-label venlafaxine, a serotonin-norepinephrine reuptake inhibitor (≤ 300 mg/d). We combined transcranial magnetic stimulation of the left motor cortex with electromyography recordings from the right hand to measure cortical inhibition using contralateral cortical silent period and paired-pulse short-interval intracortical inhibition paradigms; cortical facilitation using a paired-pulse intracortical facilitation paradigm; and short-term cortical plasticity using a paired associative stimulation paradigm. All measures were collected at baseline, 1 week into treatment (n = 23) and after approximately 12 weeks of treatment.
Results Venlafaxine did not significantly alter cortical inhibition, facilitation or plasticity after 1 or 12 weeks of treatment. Improvements in depressive symptoms during treatment were not associated with changes in cortical physiology.
Limitations The results presented here are specific to the motor cortex. Future work should investigate whether these findings extend to cortical areas more closely associated with depression, such as the dorsolateral prefrontal cortex.
Conclusion These findings suggest that antidepressant treatment with venlafaxine does not exert meaningful changes in motor cortical inhibition or plasticity in late-life depression. The absence of changes in motor cortical physiology, alongside improvements in depressive symptoms, suggests that age-related changes may play a role in previously identified abnormalities in motor cortical processes in late-life depression, and that venlafaxine treatment does not target these abnormalities.
Introduction
Late-life depression (LLD), encompassing both early-onset recurrent depression and late-onset depression in older adults over the age of 60 years, is associated with a poor prognosis.1 A substantial number of older adults with LLD do not respond to traditional oral antidepressant pharmacotherapy.2,3 Improved understanding of the neural correlates that underlie response and nonresponse to antidepressant treatment may aid in the advancement of pharmacologic treatment strategies for LLD. In particular, LLD involves a complex interplay between depression and aging,4 yet it remains unclear how cortical mechanisms that have been implicated in both depression and aging are involved in the pharmacological and therapeutic actions of antidepressants in LLD.
Several cross-sectional studies have demonstrated dampened γ-aminobutyric acid (GABA) receptor–mediated cortical inhibition in major depressive disorder (MDD) and in advancing age. Similarly, there is evidence that cortical plasticity, mediated by N-methyl-d-aspartate receptors, is reduced in both MDD and healthy aging. Cortical inhibitory or excitatory neurotransmission and long-term potentiation (LTP)–like plasticity can be noninvasively measured in humans using various transcranial magnetic stimulation (TMS) paradigms applied to the motor cortex. Such TMS measures in the motor cortex can be used to assay global cortical function, given the similar cellular structure across cortical regions.5 For example, TMS studies have provided in vivo evidence of impaired cortical inhibition and plasticity in younger and mid-life adults with depression6,7 and in older healthy adults.8–10 We have previously shown that similar abnormalities in cortical physiology in patients with early- and late-onset LLD may be influenced by depression- or aging-related changes in these same cortical processes;10 however, the relative influences of depression versus aging on the pathophysiology of depression in older adults cannot be teased apart in cross-sectional studies.
Longitudinal studies that examine the effects of antidepressant treatment on cortical inhibition and plasticity are needed to disentangle the influences of depression versus aging on LLD pathophysiology, and to clarify antidepressant mechanisms of action in LLD. In patients with mid-life MDD, magnetic resonance spectroscopy studies have demonstrated increases in cortical GABA concentrations with effective antidepressant treatment,11 and an association between increases in GABA levels and greater clinical response to antidepressant treatment.12 Although inconsistent findings have been reported,13 these findings suggest a specific link between cortical GABA levels and depressive symptoms, but these findings have not yet been extended to older adults with MDD. Moreover, magnetic resonance spectroscopy evidence in humans is limited to cortical neurotransmitter levels, rather than the functioning of cortical circuits mediated by these neurotransmitters.
Evidence from TMS studies suggests that antidepressants targeting the monoaminergic system can indirectly influence neurotransmission mediated by GABA and glutamate, and synaptic plasticity. For example, a single dose of a selective serotonin reuptake inhibitor (SSRI) or norepinephrine reuptake inhibitor (NRI) can alter TMS measures of cortical inhibition or facilitation14–17 and LTP-like cortical plasticity.18,19 Fewer TMS studies have examined the longitudinal effects of antidepressant treatment on cortical excitability. One recent study found diminished cortical inhibition after 3 months of SSRI treatment in 16 mid-life adults with depression.20 However, studies conducted to date have involved modest sample sizes, precluding the investigation of treatment response subgroups, so the relationship between antidepressant treatment response and changes in cortical physiology remains unclear. Moreover, the effects of antidepressant treatment over time on cortical inhibition, facilitation and plasticity have not yet been studied in older adults with MDD.
We used TMS to investigate changes in cortical inhibition, facilitation and plasticity during a 12-week, open-label trial of venlafaxine treatment in older adults with LLD. Venlafaxine is a commonly prescribed antidepressant for LLD,21 which acts as an SSRI at lower doses, and as both an SRI and an NRI (SNRI) at higher doses.22 We collected TMS measures pre-treatment, 1 week into treatment and after approximately 12 weeks of treatment to assess changes in cortical physiology in treatment response subgroups and to characterize the early and late pharmacological effects of venlafaxine on cortical physiology. We hypothesized that cortical inhibition, facilitation and plasticity would be altered at 12 weeks in treatment responders, but not in nonresponders.
Methods
Participants
Patients were enrolled in the Incomplete Response in Late-Life Depression: Getting to Remission (IRL-GRey) study, which consisted of 2 sequential clinical trials at the Centre for Addiction and Mental Health (ClinicalTrials.gov identifiers NCT00892047 and NCT02263248). Both clinical trials included a 12-week lead-in phase with open-label venlafaxine XR. All TMS measures were collected during this venlafaxine lead-in phase in 1 of the 2 IRL-GRey clinical trials.
All participants were 60 years of age or older; they met criteria for a diagnosis of MDD (i.e., early- or late-onset non-bipolar major depression), as established with the Structured Clinical Interview for DSM-IV;23 and they had a Montgomery–Åsberg Depression Rating Scale (MADRS)24 score of 15 or higher, reflecting moderate to severe depression. Exclusion criteria included anticonvulsant use and a comorbid neurologic, neurocognitive or psychiatric disorder other than an anxiety disorder. A high degree of suspicion of dementia was also an exclusion criterion, based on medical records, the assessment of a geriatric psychiatrist and a Mini-Mental State Examination25 score of less than 24 or a Modified Mini-Mental State Examination26 score of less than 84. The presence of vascular depression, which may differ in clinical presentation and etiology from nonvascular LLD,27,28 was not formally evaluated; thus, some patients with subtle cognitive impairment related to vascular depression may have been included. All participants provided written informed consent. This study was approved by the Centre for Addiction and Mental Health Research Ethics Board, in accordance with the Declaration of Helsinki.
Intervention
The clinical trial lead-in phase consisting of 12 weeks of open-label venlafaxine XR treatment has been described in detail elsewhere.29 The initial venlafaxine dose was 37.5 mg/d. Unless limited by adverse effects, the dose was titrated in 37.5 mg increments (approximately every 3 days) to 150 mg/d within the first 2 weeks. If remission of symptoms (MADRS score ≤ 10 for 2 consecutive visits) was not achieved after 6 weeks of treatment, the dose was titrated in 37.5 mg to 75 mg increments to a maximum of 300 mg/d or the highest tolerated dose. Patients were followed for at least 4 weeks at 300 mg/d or the maximum tolerated dose. As needed for sleep and anxiety, low doses of certain anxiolytics/sedatives were permitted during the trial, including benzodiazepines (≤ 2 mg/d of lorazepam, or equivalent), zopiclone (≤ 15 mg/d) and trazodone (≤ 50 mg/d). We performed sensitivity analyses that excluded patients on low doses of anxiolytics/sedatives during the study (Appendix 1, Table S2, available at jpn.ca/200001-a1). We used MADRS scores as the primary clinical outcome measure. Response to treatment was defined as a ≥ 50% reduction in the total MADRS score from baseline.30
Transcranial magnetic stimulation
All TMS measures were collected immediately before and after 12 weeks of venlafaxine treatment. In a subset of participants, TMS measures were also collected 1 week into treatment to assess early pharmacological effects of venlafaxine on TMS measures of cortical physiology, before substantial therapeutic effects of venlafaxine had manifested.
The TMS paradigms were conducted as described previously. 31 Briefly, we recorded the electromyography signal from the abductor pollicis brevis muscle of the right hand, and concurrently administered TMS to the left motor cortex. The TMS coil was positioned over the left motor cortex to evoke the maximum motor response in the right abductor pollicis brevis, and to induce a posterior–anterior current in the cortex. A figure-8 coil delivered monophasic pulses using 2 Magstim 200 stimulators connected with a BiStim module (Magstim). The electromyography signal was amplified (× 1000; Intronix Technologies Corporation Model 2024F), filtered (band pass 2–2.5 kHz), and digitized at 5 kHz (Micro 1401). Patients were instructed to keep their right hand relaxed, and this was verified through continuous monitoring of the electromyography recordings during the experiment. During offline analyses, all trials were checked for noise, and any trials with voluntary electromyography activity preceding the motor evoked potential (MEP) were excluded from analysis.
Resting motor threshold was first determined as the minimum stimulator intensity that elicits ≥ 50 μV MEP amplitude for 5 of 10 TMS pulses.32 The TMS test pulse intensity was then established as the stimulator intensity needed to elicit a 1 mV average peak-to-peak MEP amplitude. Short-interval intracortical inhibition (SICI) and intracortical facilitation (ICF) paradigms consisted of unconditioned and conditioned trials. In unconditioned trials, a single test pulse was administered (12 trials), and in conditioned trials, a subthreshold TMS pulse of 80% resting motor threshold preceded the test pulse by 2 ms (SICI) or 10 ms (ICF; 12 trials each). The extent of attenuation (SICI) or potentiation (ICF) of the MEP amplitude in conditioned trials was calculated as the ratio of average conditioned MEP amplitudes to average unconditioned MEP amplitudes. A subset of patients also underwent SICI with 4 ms between TMS pulses, and ICF with 15 ms and 20 ms between pulses (see Appendix 1). Next, the cortical silent period paradigm was performed. Participants first exerted maximum force on a gauge metre using a pinch grip with their thumb and index fingers. During the cortical silent period paradigm, participants then maintained contraction of the abductor pollicis brevis muscle at 20% of maximum force, which was monitored using the gauge metre. A TMS pulse of 140% resting motor threshold, consistent with previous studies of cortical silent period in depression,6,33,34 was delivered during tonic muscle contraction (10 trials). The average duration of the resulting silent period was calculated from MEP onset to the return of any electromyography activity. All TMS trials were manually checked and excluded if excess noise was identified in the electromyography signal.
Finally, in paired associative stimulation (PAS), TMS test pulses were repeatedly paired with right median nerve stimulation with a 25 ms interstimulus interval (180 trials). The intensity of the peripheral nerve stimulation was established as 3 times each participant’s sensory threshold (intensity at which the stimulation is barely perceptible). Participants were instructed to focus their attention on the stimulated hand during PAS35 and to keep a running count of the peripheral nerve stimulations. To measure attention levels, their count was periodically recorded and compared with the real count, as described previously.31 Single TMS test pulses were delivered immediately before PAS, immediately after PAS, and 15, 30 and 60 minutes after PAS (20 trials per time point). The MEP amplitudes were averaged at each time point. The PAS-induced potentiation of cortical excitability was calculated as the maximum post-PAS average MEP amplitude,36 normalized to the pre-PAS average MEP amplitude.
Statistical analyses
For each TMS measure, primary analyses consisted of a 2 × 2 analysis of covariance (ANCOVA) with a within-subject factor of time (pre- and post-treatment), a between-subject factor of treatment response (responders and nonresponders) and a time-varying covariate of resting motor threshold. The PAS attention score was also included as a time-varying covariate in the PAS analysis. The distributions of the residuals were checked using histograms and quantile–quantile (Q–Q) plots, and the ratio of the group variances was computed to assess for homogeneity of variances. Any potential outliers identified using Cook’s distance were excluded in sensitivity analyses.
To assess early pharmacological effects of venlafaxine, we compared TMS measures at baseline and week 1 using 2-tailed paired t tests. To further assess the relationship between clinical improvement and changes in cortical physiology, we performed partial correlations between change in MADRS scores and change in SICI, ICF, cortical silent period duration and maximum PAS-induced plasticity, controlling for pre- and post-treatment change in resting motor threshold. With Bonferroni correction for multiple comparisons, the significance level for each analysis was set at p < 0.013 (0.05/4). All descriptive statistics are reported as mean ± standard deviation, unless otherwise stated.
To explore whether any changes in cortical physiology at 12 weeks reflected normalization toward healthy control values, we compared post-treatment TMS measures of LLD patients with baseline TMS measures of older and younger healthy adults previously published by our group (Appendix 1).10 We also performed subgroup analyses to assess the robustness of the primary findings in subgroups of interest, including females, males, participants with early-onset depression (onset of first depressive episode before age 60), participants with late-onset depression (first onset at age 60 or later), participants previously treated with an adequate antidepressant trial, participants not previously treated with an adequate antidepressant trial, participants whose final venlafaxine dosage was less than 225 mg/d, and participants whose final venlafaxine dosage was 225 mg/d or more22,37 (Appendix 1, Table S1). Similarly, we performed sensitivity analyses to assess the robustness of the primary findings in the absence of potential confounds: left-handed38 participants or those whose handedness was unknown, participants with concurrent benzodiazepine or zopiclone use, participants taking a low dose of trazodone or another antidepressant at the time of baseline measurements due to cross-titration, or participants with a comorbid anxiety disorder (Appendix 1, Table S2). Finally, given that we observed variable responses to the TMS paradigms, we also conducted the primary analyses in only those participants who responded to the SICI paradigm (normalized MEP amplitude < 1), the ICF paradigm (normalized MEP amplitude > 1) and the PAS paradigm (maximum normalized MEP amplitude > 1; Appendix 1, Table S3). Consistent with the primary analyses, the significance threshold was defined as p < 0.013 for all exploratory subgroup analyses.
Results
Sixty-eight participants completed TMS testing before and after 12 weeks of venlafaxine treatment (41 females and 27 males; age 66.8 ± 5.7 years [range 60–86 years]; Table 1). A subset of 23 participants also completed TMS testing after 1 week of treatment (15 females and 8 males; age 67.1 ± 5.2 years). Figure 1 illustrates the final sample sizes at week 1 and week 12 for each measure, after exclusions because of voluntary electromyography activity during recordings or missing data either pre- or post-treatment. After 12 weeks of venlafaxine treatment, 29 participants were classified as responders (decrease in MADRS score 81.2 ± 16.3%), and 39 were classified as nonresponders (decrease in MADRS score 15.0 ± 23.9%). The mean venlafaxine dosage was 97.8 ± 24.6 mg/d (range 75–150 mg/d) at week 1, and 248.7 ± 72.1 mg/d (range 112.5–300 mg/d) at week 12.

Diagram of the final sample sizes 1 week and 12 weeks into venlafaxine treatment for each transcranial magnetic stimulation (TMS) measure, after data exclusions owing to voluntary movement-related noise in the electromyography signal or missing data at 1 time point. ICF = intracortical facilitation; PAS = paired associative stimulation; SICI = short-interval intracortical inhibition.
Demographic and clinical information for patients with late-life depression in treatment response subgroups
We observed no significant main effects of time and no significant time × response interactions for any of the TMS measures. In other words, 12 weeks of treatment did not lead to significant changes in cortical physiology in the whole sample, or differentially in responders and nonresponders. The descriptive statistics and ANCOVA results are summarized in Table 2, and individual patient TMS data are illustrated in Figure 2.
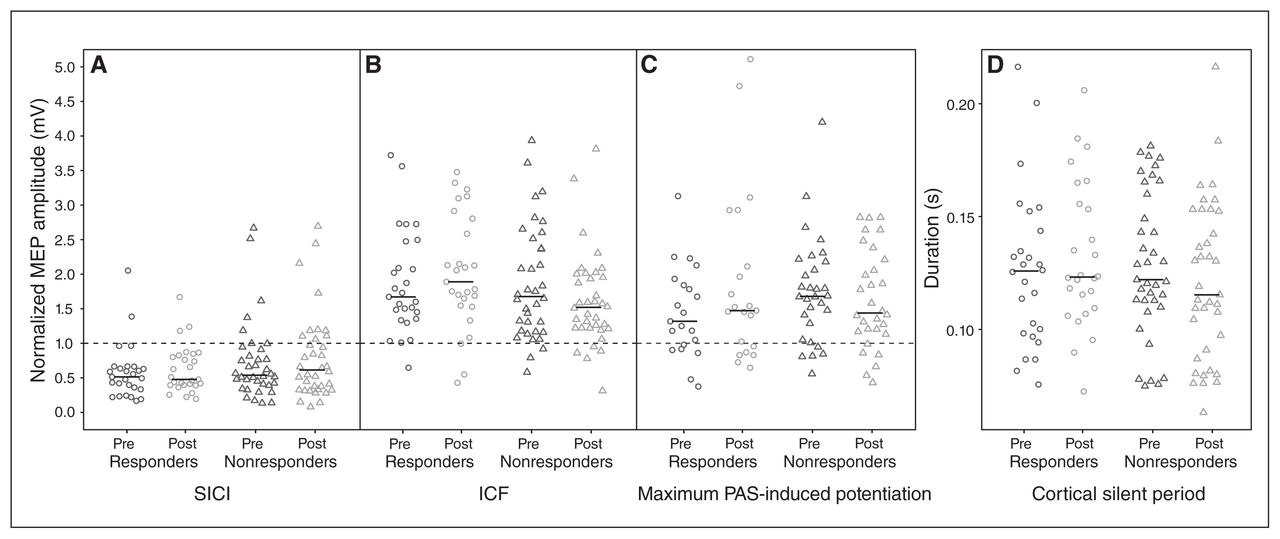
Pre- and post-treatment individual patient data by treatment response subgroup. The horizontal black bar represents the median. (A) Cortical inhibition: for the short-interval intracortical inhibition (SICI) paradigm, conditioned motor evoked potential (MEP) amplitudes (2 ms interstimulus interval) normalized to unconditioned MEP amplitudes (test pulse alone) are shown. Lower values reflect stronger inhibition. (B) Cortical facilitation: for intracortical facilitation (ICF), conditioned MEP amplitudes (10 ms interstimulus interval) normalized to unconditioned MEP amplitudes are shown. Higher values reflect greater facilitation. (C) Cortical long-term potentiation (LTP)–like plasticity: for the paired associative stimulation (PAS) paradigm, maximum post-PAS MEP amplitudes normalized to pre-PAS MEP amplitudes are shown. Higher values reflect greater PAS-induced potentiation. (D) Cortical inhibition: cortical silent period durations are shown. Longer durations reflect greater inhibition. For A, B and C, the horizontal dashed line indicates a normalized MEP amplitude of 1.0, which reflects no inhibition or facilitation of the MEP.
Descriptive statistics pre- and post-treatment with venlafaxine, pre-/post-treatment changes in EMM, and ANCOVA results for each TMS measure
We observed no significant changes in cortical physiology 1 week into treatment. We did observe a mean decrease in cortical silent period duration 1 week into treatment (baseline = 123.6 ± 30.0 ms; week 1 = 112.3 ± 28.8 ms; t22 = 2.27, p = 0.033), but the change was not significant after correction for multiple comparisons, and we observed no significant change in cortical silent period pre-/post-treatment (t62 = 0.49, p = 0.63; Table 2). We also observed no significant partial correlations between pre-/post-treatment changes in depressive symptoms and pre-/post-treatment changes in cortical silent period (r = 0.11, p = 0.41), SICI (r = 0.11, p = 0.42), ICF (r = 0.20, p = 0.11) or maximum PAS-induced plasticity (r = 0.08, p = 0.56).
Mean resting motor threshold was 48.2 ± 8.9% maximum stimulator output at baseline and 45.9 ± 9.0% after 12 weeks of treatment. Mean test pulse intensity was 62.7 ± 15.0% maximum stimulator output at baseline and 57.6 ± 13.8% after 12 weeks of treatment. The peak-to-peak MEP amplitude in response to the test pulse was 0.97 ± 0.48 mV at baseline and 1.05 ± 0.44 mV post-treatment.
Comparisons of post-treatment measures in LLD patients and baseline measures in older and younger healthy controls10 revealed reduced SICI in LLD patients compared with younger healthy adults after treatment, both in the whole sample and in treatment responders (Appendix 1). Furthermore, the negative findings reported above were consistent across all relevant subgroups, in all sensitivity analyses (Appendix 1, Tables S1 to S3) and using a wider range of paired-pulse interstimulus intervals (Appendix 1). When including only participants who responded to SICI, ICF and PAS paradigms, interaction and main effects remained nonsignificant (Appendix 1, Table S3). Finally, all findings remained nonsignificant when potential outliers identified with Cook’s distance were excluded.
Discussion
To our knowledge, this was the first investigation of changes in cortical inhibition, facilitation and plasticity with antidepressant treatment in older adults with MDD. We used the motor cortex as a proxy for neurophysiological processes occurring across other cortical regions, based on similar canonical microcircuits across the cortex.5 Although regional variability may occur based on receptor densities, the pharmacological effects of venlafaxine are also likely to occur across the cortex, because serotonin and norepinephrine transporters are distributed across the cortex.39,40 The response to venlafaxine treatment observed here (42.6% response, 57.4% nonresponse) was consistent with previously reported venlafaxine response rates in LLD3 and resulted in large enough samples of responders and nonresponders to investigate changes in cortical physiology based on individual therapeutic responses to treatment. Contrary to our hypotheses, therapeutic response to 12 weeks of venlafaxine treatment was not associated with significant pre-/post-treatment changes in cortical inhibition, facilitation or plasticity. Similarly, we observed no significant SNRI pharmacology-related changes in cortical physiology 1 week or 12 weeks into treatment. These results were robust in subgroup analyses that considered the influence of sex, MDD age of onset, history of treatment resistance, venlafaxine dosage, handedness, concurrent drug use, comorbid anxiety disorder and TMS paradigm outliers.
The current findings highlight the influence that aging may have on the neurobiology of depression in older adults41 and the mechanisms by which antidepressants may exert their effects in older adults. A repeated-measures study design allowed us to assess the relationship between changing depressive states and cortical physiology in LLD, while age remained almost constant. Cortical inhibition/facilitation and plasticity did not change alongside improvements in depressive symptoms with treatment. Furthermore, previously identified deficits in cortical inhibition in LLD patients compared with younger healthy adults10 persisted after effective treatment (see Appendix 1). Taken together, these findings suggest that venlafaxine treatment does not target abnormalities in cortical physiology in LLD. Additionally, given accumulating evidence in healthy adults across the lifespan that cortical inhibition declines with advancing age,8,10,42,43 abnormalities in cortical inhibition may be more markedly influenced by aging than by depressive states in LLD. These observations support the age × disease interaction hypothesis of LLD,4 which postulates that the biological changes that occur during aging overlap with those involved in the pathology of depression and other age-gated diseases. Alternatively, given the elevated risk of developing dementia in LLD,44 abnormalities in cortical physiology in LLD may be more a manifestation of insidious neurodegenerative processes that occur with vascular-related cognitive deterioration.45 The contribution of age or LLD-related neurodegenerative processes to persisting deficits in cortical functioning could, for instance, contribute to the chronicity of depressive symptoms,46 high relapse rates after treatment47 or the persistence of cognitive symptoms following antidepressant treatment,48 all of which have been observed in LLD. Therefore, novel forms of treatment that target cortical functioning49 may be beneficial for some LLD patients.
The current findings were surprising in light of previous evidence that cortical inhibition, facilitation and LTP-like plasticity may change with other forms of treatment, including alternative medicine50–52 and brain stimulation treatment.53–56 Accordingly, alternative treatments such as brain stimulation may involve different biological mechanisms,57 and the current findings may therefore be specific to venlafaxine pharmacotherapy. However, previous studies focused on younger and mid-life adults with depression, and did not account for the added influence of advancing age on cortical physiology.
The current findings are also in contrast to previous those of studies in small samples of mid-life adults with depression that showed normalization of cortical GABA and glutamate concentrations with effective antidepressant pharmacotherapy. 11,58 Although age may play a role in our observation that certain motor cortical processes do not undergo meaningful changes with antidepressant treatment, other cortical regions and molecular mechanisms involved in inhibition/facilitation or plasticity might be involved in antidepressant treatment response. For example, a recent TMS study found that 3 months of escitalopram treatment led to changes in long-interval cortical inhibition, but not SICI or ICF, in 16 adults with depression in mid-life.20 Long-interval cortical inhibition is thought to specifically reflect GABA-B receptor–mediated neurotransmission, whereas SICI reflects GABA-A receptor–mediated cortical inhibition, 59 and the cortical silent period may reflect both GABA-A and GABA-B receptor–mediated activity.59 Therefore, it is possible that cortical inhibition selectively mediated by GABA-B receptors changes with venlafaxine treatment, although this possibility would need to be investigated in larger samples of patients, and in LLD patients in particular.
Similarly, the absence of pharmacological effects of venlafaxine on cortical inhibition, facilitation or plasticity in patients with LLD contrasted with earlier findings of changes in TMS measures that followed known drug pharmacology, such as the reliable lengthening of the silent period following clozapine treatment.60 Accumulating evidence from rodent and magnetic resonance spectroscopy studies suggests that monoaminergic antidepressants indirectly influence GABA, glutamate and synaptic plasticity.11,61–64 Studies of TMS in healthy adults have also shown elevated cortical facilitation65 and reduced cortical inhibition66 following chronic SSRI or NRI administration. Here, the unique pharmacology of venlafaxine as an SNRI may have contributed to interindividual variability in cortical physiology changes with treatment. For example, previous TMS studies have found that an acute dose of a selective NRI increases cortical facilitation and decreases cortical inhibition,17,66 whereas an acute dose of an SSRI decreases cortical facilitation and increases cortical inhibition. 16 However, with venlafaxine, the level of inhibition of norepinephrine uptake is dose-dependent,22,37 and the findings remained nonsignificant in participants who took a low final dosage of venlafaxine (i.e., < 225 mg/day), at which venlafaxine is expected to act as an SSRI (see Appendix 1, Table S1). Alternatively, interindividual variability in the effects of venlafaxine on cortical excitability could be accounted for by other factors such as genetic polymorphisms.67
Limitations
Our findings should be considered in light of several limitations. First, although cortical regions are similar in composition, the current findings are specific to the motor cortex and may not apply to other cortical areas implicated in depression. Future work should investigate changes in cortical physiology in the dorsolateral prefrontal cortex with treatment in LLD. Second, the majority of the LLD participants were concurrently medicated during the trial, and had previously failed to respond to at least 1 adequate antidepressant trial. Subgroup analyses excluding participants with a history of treatment resistance or concurrent benzodiazepine, zopiclone or antidepressant use suggest that treatment resistance and concurrent drug use do not account for the observed negative findings. However, this possibility cannot be ruled out. Third, TMS measures of cortical inhibition, facilitation and plasticity constitute indirect measures of these cortical processes. Other modalities, such as magnetic resonance spectroscopy, would complement the current TMS investigation of changes in GABAergic and glutamatergic processes with LLD treatment. Fourth, the current study focused on TMS measures from the left hemisphere that have previously been implicated in depression pathophysiology,6,10,33,34,68 but future work should assess whether changes in other TMS measures — such as interhemispheric or bilateral measures or the MEP/compound muscle action potential amplitude ratio — occur during venlafaxine treatment. Fifth, a formal diagnostic assessment of dementia was not part of the neurocognitive assessment, so a diagnosis of dementia based on DSM-5 criteria cannot be excluded. Finally, we did not perform a comprehensive assessment for vascular depression, so some patients with vascular depression may have been included in the study. Although the primary findings were robust in subgroup analyses of only early-onset or late-onset patients, the pathophysiology of late-onset vascular depression and early-onset recurrent depression may nevertheless differ.28,69 As such, future work in a larger sample of late-onset LLD patients, including diagnoses of vascular depression based on structural/functional neuroimaging and a neuropsychological battery, is merited.
Conclusion
Our findings suggest that the therapeutic and pharmacological actions of venlafaxine do not involve changes in motor cortical inhibition, facilitation or LTP-like plasticity in older adults with depression. Given that these cortical processes have been previously linked to both advancing age and depression, the observed stability of the TMS measures over time despite treatment-related depressive symptom improvement suggests that age-related changes may be an important factor driving motor cortex physiologic functioning. Future TMS work should include both younger and older adults with depression and leverage electroencephalography measurements of cortical output to examine whether age- and depression-linked cortical processes change with antidepressant treatment in other cortical regions.
Footnotes
Funding: This work was directly supported by NIMH grant NIH R34MH101365.
Competing interests: B. Mulsant reports non-financial support from Capital Solution Design LLC, HAPPYneuron, Bristol-Myers Squibb and Eli Lilly; grants from Brain Canada, the Patient-Centered Outcomes Research Institute (PCORI), the Canadian Institutes of Health Research (CIHR), the National Institutes of Health (NIH) and the Centre for Addiction and Mental Health (CAMH) Foundation; and other support from General Electric. T. Rajji has received research support from Brain Canada, the Brain and Behavior Research Foundation, the BrightFocus Foundation, the Canada Foundation for Innovation, a Canada Research Chair, CIHR, the Centre for Aging and Brain Health Innovation, NIH, the Ontario Ministry of Health and Long-Term Care, the Ontario Ministry of Research and Innovation and the Weston Brain Institute; in-kind equipment support for an investigator-initiated study from Magstim; and in-kind research accounts from Scientific Brain Training Pro. J. Karp is a scientific advisor to NightWare and received medication supplies for investigator-initiated trials from Pfizer and Indivior and honoraria for development and presentation of a webinar (disease-state, not product-focused) from Otsuka. C. Reynolds III reports grants from the NIH during the conduct of the study; grants from the NIH, the PCORI, the Center for Medicare and Medicaid Services, the American Foundation for Suicide Prevention, the Brain and Behavior Research Foundation and the Commonwealth of Pennsylvania outside the submitted work; and non-financial support from Bristol Meyers Squib and Pfizer, outside the submitted work. E. Lenze received research support from the National Institute on Aging, the National Center for Complementary and Integrative Health, the National Institute of Mental Health ( NIMH), the Office of Behavioral and Social Sciences Research, the United States Food and Drug Administration, PCORI, the McKnight Brain Research Foundation, the Taylor Family Institute for Innovative Psychiatric Research, the Barnes Jewish Foundation, Takeda, Acadia Alkermes, Aptinyx and Lundbeck; and consulting fees from Janssen and Jazz Pharmaceuticals. J. Downar reports grants from the Arrell Family Foundation, the Buchan Family Foundation, Brain Canada, the Canadian Biomarker Integration Network in Depression, CIHR, the Klarman Family Foundation, NIMH, the Ontario Brain Institute and the Weston Family Foundation; and other support from ANT Neuro, BrainCheck, Restorative Brain Clinics and TMS Neuro Solutions, all outside the submitted work. R. Chen received honoraria from Allergan, Merz and Ipsen. Z. Daskalakis reports non-financial support from Brainsway Inc; non-financial support from Magventure Inc; grants from the Ontario Mental Health Foundation, CIHR, NIMH, the Temerty Family, the Grant Family, the CAMH Foundation and the Campbell Institute, all outside the submitted work. D. Blumberger has received research support from CIHR, NIMH, Brain Canada and the Temerty Family through the CAMH Foundation and the Campbell Family Research Institute; received research support and in-kind equipment support for an investigator-initiated study from Brainsway Ltd; is the site principal investigator for one sponsor-initiated study for Brainsway Ltd.; receives in-kind equipment support from Magventure for investigator-initiated research; and received medication supplies for an investigator-initiated trial from Indivior. No other competing interests were declared.
Contributors: B. Mulsant, T. Rajji, C. Reynolds III, E. Lenze, Z. Daskalakis and D. Blumberger designed the study. J. Lissemore, B. Mulsant, J. Karp, E. Lenze, Z. Daskalakis and D. Blumberger acquired the data, which J. Lissemore, B. Mulsant, T. Rajji, C. Reynolds III, E. Lenze, J. Downar, R. Chen, Z. Daskalakis and D. Blumberger analyzed. J. Lissemore, E. Lenze and D. Blumberger wrote the article, which all authors reviewed. All authors approved the final version to be published and can certify that no other individuals not listed as authors have made substantial contributions to the paper.
- Received January 3, 2020.
- Revision received May 30, 2020.
- Accepted June 18, 2020.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is non-commercial (i.e. research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/
References
In this issue

{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.