
Abstract
Converging evidence implicates immune abnormalities in schizophrenia (SCZ), and recent genome-wide association studies (GWAS) have identified immune-related single-nucleotide polymorphisms (SNPs) associated with SCZ. Using the conditional false discovery rate (FDR) approach, we evaluated pleiotropy in SNPs associated with SCZ (n=21 856) and multiple sclerosis (MS) (n=43 879), an inflammatory, demyelinating disease of the central nervous system. Because SCZ and bipolar disorder (BD) show substantial clinical and genetic overlap, we also investigated pleiotropy between BD (n=16 731) and MS. We found significant genetic overlap between SCZ and MS and identified 21 independent loci associated with SCZ, conditioned on association with MS. This enrichment was driven by the major histocompatibility complex (MHC). Importantly, we detected the involvement of the same human leukocyte antigen (HLA) alleles in both SCZ and MS, but with an opposite directionality of effect of associated HLA alleles (that is, MS risk alleles were associated with decreased SCZ risk). In contrast, we found no genetic overlap between BD and MS. Considered together, our findings demonstrate genetic pleiotropy between SCZ and MS and suggest that the MHC signals may differentiate SCZ from BD susceptibility.
Similar content being viewed by others
Introduction
Schizophrenia (SCZ) and Bipolar Disorder (BD) are severe mental disorders, which are among the leading causes of disability globally.1 These disorders have a substantial impact on the quality of life for patients and their families, and are among the most costly societal disorders.2 Clinical, epidemiological and genetic findings suggest shared risk factors between BD and SCZ, and a Psychosis Continuum Model has been suggested.3 Despite the high heritability, most of the genetic architecture underlying susceptibility to both SCZ and BD remains to be defined, and the pathobiological mechanisms underlying these disorders are still largely unknown. Improved understanding of disease pathobiology and genetic risk factors may lead to major health benefits through the development of new treatment and prevention regimens.4
An interesting hypothesis regarding SCZ pathology is the involvement of the immune system, which is derived from epidemiological and clinical evidence, implicating infections5 and cytokine abnormalities6 in the development of SCZ. Recent genome-wide association studies (GWAS) have robustly identified markers in the major histocompatibility complex (MHC) associated with SCZ,7, 8, 9 also showing association of specific human leukocyte antigen (HLA) alleles.10 Although this may further support a role of the immune system in SCZ pathogenesis, it is also possible that this association reflects nonimmunological factors as several loci within the MHC are involved in neurobiological processes (for example, NOTCH4, TRIM26).11 With regard to BD, despite a large degree of overlapping clinical characteristics, genetic factors as well as cytokine abnormalities6,12 between BD and SCZ, there is little evidence for MHC associations in GWAS of BD.13 Our previous analysis of the shared genetic basis of these disorders revealed significant genetic overlap, but no common loci in the MHC.14 Together, this suggests differences in MHC-related genes between the two disorders.
Multiple sclerosis (MS) is a disease of the central nervous system in which inflammatory processes have an important role and associations with HLA class I and II loci are well established.15 The HLA-DRB1*1501 allele shows strong association with MS susceptibility in most populations with an average odds ratio (OR) of 3.16 A recent GWAS in MS identified more than 50 nonMHC regions associated with MS risk, and immunologically relevant genes were significantly over-represented in these regions.16 Further studies and meta-analyses have increased the number of detected MS risk variants.17, 18, 19 The immunological mechanisms involved in MS leads to various degrees of demyelination,20 and several lines of evidence point to oligodendrocyte and myelin dysfunction also in SCZ.21 This suggests shared risk factors between MS and SCZ. Given the overlapping phenotypes and polygenic architecture between SCZ and BD, genetic pleiotropy between BD and MS may also exist.
Combining GWAS from two disorders provides increased power to detect significant associations, and recent studies have reported overlapping single-nucleotide proteins (SNPs) between several human traits22,23 and disorders.13,23 Here, we use MS GWAS data from the International MS Genetics Consortium (IMSGC)16 together with SCZ GWAS7 and BD GWAS13 data to investigate pleiotropic relationships between these disorders. To date, methods to assess genetic pleiotropy have not taken full advantage of the existing GWAS data, and the majority of these studies have focused on the subset of SNPs exceeding a Bonferroni-corrected threshold of significance for each trait or disorder.7,23 However, using this approach, SNPs with nonsignificant effects in each phenotype, but significant in the combined analysis (polygenic pleiotropy), cannot be detected. We recently developed a novel analytic strategy14,24,25 that uses all available SNPs in two or more independent GWAS to identify polygenic pleiotropy and improve gene discovery. Using this approach, we demonstrated extensive polygenic pleiotropy between SCZ and BD,14 and between SCZ and cardiovascular disease risk factors (CVD),24 and substantially increased the number of identified SCZ genetic susceptibility loci.14,24,25 Here, we applied these methods to independent large MS, SCZ and BD GWAS to determine whether MS shares susceptibility loci with these psychiatric disorders.
Materials and methods
Participant samples
We used summary statistics from a large MS GWAS study performed by IMSGC,16 n=27 148, and from two large GWAS studies from the Psychiatric GWAS Consortium (PGC), PGC Schizophrenia sample,7 n=21 856, PGC Bipolar disorder sample,13 n=16 731. P-values and minor allele frequencies from the discovery samples were included in the analyses. For follow-up analysis, we also investigated the PGC major depressive disorder,26 autism spectrum disorder27 and attention deficit/hyperactivity disorder28 GWAS summary statistics (for additional details, see Supplementary Information).
Statistical analyses
Conditional Q-Q plots for pleiotropic enrichment
To visually assess pleiotropic enrichment, we used Q-Q plots conditioned on ‘pleiotropic’ effects14,24 (Figures 1a and 2a for BD). For a given associated phenotype, pleiotropic ‘enrichment’ exists if the degree of deflection from the expected null line is dependent on associations with the second phenotype (for further details see Supplementary Information). We constructed conditional Q-Q plots of empirical quantiles of nominal –log10(P) values for all SNPs and for subsets of SNPs determined by the significance of their association with MS. Specifically, we computed the empirical cumulative distribution function (ecdf) of nominal P-values for a given phenotype for all SNPs and for SNPs with significance levels below the indicated cutoffs for the other phenotype (–log10(P)⩾0, –log10(P)⩾1, –log10(P) ⩾2, –log10(P) ⩾3 corresponding to P⩽1, P⩽0.1, P⩽0.01, P⩽0.001, respectively). Nominal P-values (–log10(P)) are plotted on the y-axis, and empirical quantiles (–log10(q), where q=1−ecdf(P)) are plotted on the x-axis. To assess polygenic effects below the standard GWAS significance threshold, we focused the Q-Q plots on SNPs with nominal –log10(P)<7.3 (corresponding to P>5 × 10−8). The same procedure was used for BD. The ‘enrichment’ seen in the conditional Q-Q plots can be directly interpreted in terms of true discovery rate (TDR=1− FDR).29 This is illustrated in Figures 1b and 2b for each range of P-values in the pleiotropic traits (see Supplementary Information for details).

Genetic pleiotropy enrichment of schizophrenia (SCZ) conditional on MS. (a) Conditional Q-Q plot of nominal versus empirical −log10 P-values (corrected for inflation) in SCZ below the standard GWAS threshold of P<5 × 10−8 as a function of significance of association with multiple sclerosis (MS) at the level of −log10(P)⩾0, −log10(P)⩾1, −log10(P)⩾2, −log10(P)⩾3 corresponding to P⩽1, P⩽0.1, P⩽0.01, P⩽0.001, respectively. Dotted lines indicate the null hypothesis. (b) Conditional true discovery rate (TDR) plots illustrating the increase in TDR associated with increased pleiotropic enrichment in SCZ conditioned on MS (SCZ|MS). (c) Cumulative replication plot showing the average rate of replication (P<0.05) within SCZ substudies for a given P-value threshold shows that pleiotropic enriched SNP categories replicate at a higher rate in independent SCZ samples, for SCZ conditioned on MS (SCZ|MS). The vertical intercept is the overall replication rate per category. (d) Z-score-z-score plot demonstrates that the empirical replication z-scores closely match the expected a posteriori effect sizes of SCZ and are strongly dependent upon pleiotropy with MS. Analysis is based on split half method of the 17 PGC SCZ substudies.

Genetic pleiotropy enrichment of BD conditional on MS. (a) Conditional Q-Q plot of nominal versus empirical −log10 P-values (corrected for inflation) in bipolar disorder (BD) below the standard GWAS threshold of P<5 × 10−8 as a function of significance of association with multiple sclerosis (MS) at the level of −log10(P)⩾0, −log10(P)⩾1, −log10(P)⩾2, −log10(P)⩾3 corresponding to P⩽1, P⩽0.1, P⩽0.01, P⩽0.001, respectively. Dotted lines indicate the null hypothesis. (b) Conditional true discovery rate (TDR) plots illustrating the increase in TDR associated with increased pleiotropic enrichment in BD conditioned on MS (BD|MS).
Conditional replication rate
For each of the 17 substudies contributing to the final meta-analysis in SCZ, we independently adjusted the z-scores using intergenic inflation control.30 We randomly sampled 1000 combinations of eight and nine substudy groupings. We then calculated the eight-or-nine-study combined discovery z-score and eight-or-nine-study combined replication z-score for each SNP as the average z-score across the substudies multiplied by the square root of the number of studies. For discovery samples, the z-scores were converted to two-tailed P-values, whereas replication samples were converted to one-tailed P-values preserving the direction of effect in the discovery sample. For each of the 1000 discovery-replication pairs, cumulative rates of replication were computed over 1000 equally spaced bins spanning the range of −log10(P-values) observed in the discovery samples. The cumulative replication rate for any bin was the proportion of SNPs with a −log10 (discovery P-value) greater than the lower bound of the bin with a replication P-value<0.05 and the same sign as the discovery sample. Cumulative replication rates were calculated independently for each of the four pleiotropic enrichment categories. For each category, the cumulative replication rate for each bin was averaged across the 1000 discovery-replication pairs, and the results are reported in Figure 1c. The vertical intercept in the figure is the overall replication rate.
Conditional replication effect size
Using the same z-score adjustment scheme and sampling method used for estimating cumulative replication rates (see above), we directly evaluated the relationship of replication effect size of the discovery sample versus replication samples (Figure 1d) for each SNP. The effect sizes were conditioned on various enrichment categories. For visualization, we fitted a cubic spline relating the bin mid-point of z-scores of discovery to the corresponding average replication z-scores (Figure 1d).
Improving discovery of SNPs in SCZ and BD using conditional FDR
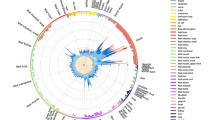
To improve detection of SNPs associated with SCZ and BD, we employed a genetic epidemiology approach, leveraging the MS phenotype from an independent GWAS using conditional FDR as outlined in Andreassen et al.14,24 Specifically, conditional FDR is defined as the posterior probability that a given SNP is null for the first phenotype given that the P-values for both phenotypes are as small as or smaller than their observed P-values. We assigned a conditional FDR value for each SNP in SCZ given the P-value in MS (denoted as FDRSCZ|MS). All SNPs with conditional FDR<0.05 (−log10(FDR)>1.3) in SCZ given the association with MS are listed in Supplementary Table 1. The same procedure was applied to compute FDRBD|MS for each SNP. To display the localization of the genetic markers associated with SCZ and BD given the MS effect, we used a ‘Conditional Manhattan plot’, plotting all SNPs within an LD block in relation to their chromosomal location. As illustrated for SCZ in Figure 3, the large points represent the significant SNPs (−log10(FDRSCZ|MS)>1.3 equivalent to FDRSCZ|MS<0.05), whereas the small points represent nonsignificant SNPs. All SNPs are shown without ‘pruning’ (that is, without removing all SNPs with r2>0.2 based on 1000 Genome Project (1KGP) linkage disequilibrium (LD) structure). The strongest signal in each LD block is illustrated with a black line around the circles. This was identified by ranking all SNPs in increasing order, based on the FDRSCZ | MS value and then removing SNPs in LD r2>0.2 with any higher-ranked SNP. Thus, the selected locus was the most significantly associated with SCZ in each LD block.

‘Conditional FDR Manhattan plot’. Conditional FDR Manhattan plot of conditional –log10 (FDR) values for schizophrenia (SCZ) alone (gray) and SCZ conditioned on multiple sclerosis (MS; SCZ|MS, red). SNPs with conditional –log10 FDR>1.3 (that is, FDR<0.05) are shown with large points. A black line around the large points indicates the most significant SNP in each LD block. This SNP is annotated with the closest gene. Genes are listed in increasing order in terms of SNPs genomic position within each chromosome from left to right and novel ones are marked by stars (*). The figure shows the localization of the 21 independent loci on a total of 13 chromosomes.
Annotation of novel loci
On the basis of 1KGP linkage disequilibrium (LD) structure, significant SNPs identified by conditional FDR were clustered into LD blocks at the LD−r2>0.2 level. These blocks are numbered (locus #) in Tables 1 and 2. Any block may contain more than one SNP. Genes close to each SNP were obtained from the NCBI gene database. Only blocks that did not contain previously reported SNPs or genes related to previously reported SNPs were deemed as novel loci in the current study (Tables 1 and 2). Loci that contained either SNPs or genes known to be associated with SCZ were considered as replication findings.
HLA allele analysis
The PGC1 genotype data from the 17 substudies were used for HLA imputation (a detailed description of the data sets, quality control procedures, imputation methods, and, principal components estimation, are given in Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium.7 First, genotypes of SNPs in the extended MHC (Major Histocompatibility Complex) (chr6: 25652429–33368333) of each individual in all the samples were extracted. Then, the program HIBAG31 was used to impute genotypes of classical HLA alleles for each sample separately, using the parameters trained on the Scottish 1958 birth cohort data (http://www.b58cgene.sgul.ac.uk/). HLA alleles with posterior probabilities ⩾0.5 and frequency >0.01 were used in subsequent analysis. The genotypes of the 63 HLA alleles meeting these criteria were encoded as binary variables for the following conditional analysis.
Samples with imputed HLA genotypes were combined before the analysis. First, the logistic regression method implemented in PLINK32 was employed to test HLA alleles for associations with SCZ, using the first five principal components and sample indicator variable as covariates. After Bonferroni correction, five alleles passed the genomic significance threshold (7.9 × 10-4, see Supplementary Table 2).
The dosages of SNPs in the MHC, imputed based on HapMap3 data, were tested using logistic regression. The analysis was first performed with only sample indicator variables and the first five principal components as covariates and then including, in turn, one of the significant HLA alleles from the previous step as an additional covariate. In addition to the SCZ-associated HLA alleles, four other alleles reported to be associated with MS were also tested in this framework. A large increase in a SNP’s association P-value upon conditioning on HLA alleles is considered to indicate overlap with that HLA allele (Supplementary Figure 5).
Results
Enrichment of SCZ SNPs due to association with MS—conditional Q-Q plots
Conditional Q-Q plots for SCZ given level of association with MS (Figure 1a) show variation in enrichment. Earlier (and steeper) departures from the null line (leftward shift) with higher levels of association with MS indicate a greater proportion of true associations (Figure 1b) for a given nominal P-value. The divergence of the curves for different conditioning subsets thus suggests that the proportion of non-null effects varies considerably across different degrees of association with MS. For example, the proportion of SNPs in the −log10(pMS) ⩾3 category reaches a given significance level (−log10(pSCZ)>6) that is roughly 50–100 times greater than for the −log10(pMS)⩾0 category (all SNPs), indicating considerable enrichment. The enrichment was significant after pruning, as shown by the Q-Q plots with confidence intervals given in Supplementary Figure 1. The enrichment also remained significant after removing the SNPs with genome-wide significant P-values (censored Q-Q plots, Supplementary Figure 2). In contrast, we found no evidence for enrichment in BD conditional on MS (Figure 2).
Association with MS increases conditional true discovery rate (TDR) in SCZ
Variation in enrichment in pleiotropic SNPs is associated with corresponding variation in conditional TDR, equivalent to one minus the conditional FDR.29 A conservative estimate of the conditional TDR for each nominal P-value is equivalent to 1–(P/q) as plotted on the conditional Q-Q plots (see Methods). This relationship is shown for SCZ conditioned on MS in a conditional TDR plot (Figure 1b; TDRSCZ | MS, and for BD Figure 2b; TDRBD|MS). For a given conditional TDR, the corresponding estimated nominal P-value threshold varied by a factor of 100 from the most to the least enriched SNP category for SCZ conditioned by MS. Since the conditional TDR is strongly related to predicted replication rate, the replication rate is expected to increase for SNPs in categories with higher conditional TDR.
Replication rate in SCZ is increased by MS association
To address the possibility that the observed pattern of differential enrichment results from spurious (that is, nongeneralizable) associations due to category-specific stratification or statistical modeling errors, we examined the empirical replication rate across independent substudies for SCZ. Figure 1c shows the empirical cumulative replication rate plots as a function of nominal P-value, for the same categories as for the conditional Q-Q and TDR plots in Figures 1a and b. Consistent with the conditional TDR pattern, we found that the nominal P-value corresponding to a wide range of replication rates was 100 times higher for −log10 (pMS) ⩾3 relative to the −log10 (pMS) ⩾0 category (Figure 1c). Similarly, SNPs from pleiotropic SNP categories showing the greatest enrichments (−log10 (pMS)⩾3) replicated at highest rates, up to five times higher than all SNPs (−log10(pMS)⩾0), for a wide range of P-value thresholds. This strongly suggests that replication of SNP associations varies as a function of estimated conditional TDR.
Replication effect size depends upon MS association
Consistent with the pattern observed for replication rates in SCZ substudies (see above), we found that the effect sizes of SNPs in enriched categories (for example, −log10 (pMS)⩾3) replicated better than effect sizes of SNPs in less-enriched categories (for example, −log10(pMS)⩾0; Figure 1d). This indicates that the fidelity of replication effect sizes is closely related to the conditional TDR.
SCZ gene loci identified with conditional FDR
Conditional FDR methods14,24 improve our ability to detect SNPs associated with SCZ due to the additional power generated by use of the MS GWAS data. Using the conditional FDR for each SNP, we constructed a ‘conditional FDR Manhattan plot’ for SCZ and MS (Figure 3). The reduced FDR obtained by leveraging association with MS enabled us to identify loci significantly (conditional FDR<0.05) associated with SCZ on a total of 13 chromosomes. We subsequently pruned the associated SNPs (removed SNP with LD−r2>0.2) and identified a total of 21 independent loci, of which one complex locus was located in the MHC on chromosome 6 (Table 2) and 20 single-gene loci were located in chromosomes 1–3, 6–12, 14, 15 and 18 (Table 1). These loci are marked by large points with black edges in Figure 3. Only ten of the independent loci have been identified by previous SCZ GWASs using standard analysis.7,10 However, several have also been identified in our previous analyses of genetic pleiotropy between SCZ and cardiovascular disease risk factors (CVD)24 and between SCZ and BD14 (Tables 1 and 2). All SNPs with conditional FDR<0.05 are listed in Supplementary Table 1.
Effect of the size of strata on enrichment
The observed enrichment was further confirmed by performing the same analysis on additional categories (−log10 (pMS)⩾4, −log10 (pMS)⩾5 and −log10 (pMS)⩾6, Supplementary Figure 3). Although the general enrichment pattern persisted, the number of valid SNPs, which exist in both SCZ and MS data set and also have valid P-values, in these extra categories was smaller. In total, 425 028 SNPs having valid P-values for both SCZ and MS were analyzed in this study. They contribute 425 028, 47410, 7077, 1781, 808, 525 and 391 to the six categories conditioned by the significance level of MS, respectively.
Distribution of allele frequencies in strata
We also investigated the distribution of the minor allele frequencies (MAF) of the corresponding SNPs of each stratum from the 1KGP. Supplementary Figure 4 shows the average MAF *(1−MAF), namely, the genetic variance, in strata after pruning SNPs in LD (r2>0.2). As the significance level of SNPs with MS increases, there is a noticeable increase in average genetic variance, which is expected as MAF confounds multiplicatively with the true effect size of the variants.30 However, the effect of MAF alone cannot explain the observed enrichment (see Supplementary Figure 4).
HLA imputation and association analysis
Among the loci identified by conditional FDR methods, eight are located in the MHC (Table 2). It is possible that these signals may be driven by common HLA alleles affecting both SCZ and MS. To test this hypothesis, we imputed HLA class I and class II alleles using the PGC1 genotype data (see Materials and Methods). We performed association analysis between imputed HLA alleles and SCZ. We replicated earlier findings10 that the alleles HLA-B*08:01, HLA-C*07:01, HLA-DRB1*03:01, HLA-DQA1*05:01 and HLA-DQB1*02:01 are negatively associated with SCZ (P<7.8 × 10−4 ). Among these, HLA-DRB1*03:01 and HLA-DQB1*02:01 have been reported to be positively associated with MS.16 However, no association was seen with SCZ for the strong MS predisposing HLA-DRB1*15:01 and HLA-DRB1*13:03 alleles, nor for the protective HLA-A*02:01 allele. We further tested whether SNPs in the MHC with conditional FDR<0.05 were independent of the association signal with the classical HLA alleles (see Materials and Methods). SNPs rs9379780, rs3857546, rs7746199, rs853676 and rs2844776 seem to be independent of the HLA allelic signal (Supplementary Figure 5 and Supplementary Table 2). We further tested if the associated HLA alleles were independent of each other by conditional analysis between them (see Materials and Methods). The results in Supplementary Table 3 suggest that the observed associations are driven by a single haplotype block (that is, ancestral haplotype 8.1), consisting of the five individual HLA alleles. However, because of the low number of successfully imputed HLA alleles, it is difficult to exclude independent signals.
The effect of MHC SNPs on enrichment
We also investigated the effect of MHC-related SNPs (SNPs located within the MHC or SNPs within 1 Mb and in LD (r2>0.2) with such SNPs) on the observed enrichment for SCZ and BD conditional on MS (see Supplementary Information and Supplementary Figure 6). After removing the MHC-related SNPs, the enrichment of SCZ conditioned on MS was substantially attenuated (Supplementary Figure 6). In contrast, removing the MHC-related SNPs did not affect the enrichment of BD conditioned on MS (Supplementary Figure 6). Further, we also investigated the effect of removing the MHC-related SNPs on the previously reported enrichment of SCZ conditioned on BD. As illustrated in Supplementary Figure 8, the enrichment between BD and SCZ was not affected by removing the MHC-related SNPs.
Enrichment analysis of other psychiatry disorders
Using the analysis approach described above, we evaluated genetic enrichment in major depressive disorder,26 autism spectrum disorder27 and attention deficit/hyperactivity disorder28 GWAS summary statistics from the PGC conditioned on MS. In contrast to SCZ, none of these phenotypes demonstrated significant enrichment (Supplementary Figure 7).
Discussion
Our results demonstrate genetic overlap between SCZ and MS and identify 21 independent loci associated with SCZ, which are driven by the overlap in the MHC region. In contrast, despite known genetic overlap between SCZ and BD, we found no overlap between BD and MS. As most of the overlap between MS and SCZ was driven by the MHC, and given the previous evidence that the genetic overlap between BD and SCZ is located outside the MHC, our findings suggest that the MHC cluster could differentiate between BD and SCZ susceptibility.
These results implicate shared molecular pathways between SCZ and MS. Many of the common association signals between SCZ and MS are located on chromosome 6, strongly suggesting the MHC, and possibly HLA alleles, in SCZ development. This is in line with previous findings of common variants in the MHC associated with SCZ,7,8,10,33 which seems to implicate immune factors. The current SNP and HLA allelic analyses indicate that there could be several risk loci in the MHC for SCZ, possibly including both classical antigen-presenting HLA alleles and other immune loci. The DRB1*03:01 and DQB1*02:01 alleles that increase risk of MS were found to decrease the risk for SCZ. Other MS-associated HLA alleles were not found to be associated with SCZ in our analysis. Thus, although MS and SCZ are both associated with several loci in the MHC, their genetic risk profile appears to be divergent. Because of the low imputation rate in our study, it is difficult to draw firm conclusions about precisely which loci and allelic variants in the MHC are involved in SCZ. Several of the nonMHC loci suggested to be involved in SCZ susceptibility by the current study are also related to immune pathways, which are established as key mechanisms in MS pathogenesis.16 Further, support for immune involvement in SCZ comes from recent findings of abnormalities in cytokine levels and other immune markers in SCZ6,34 and from strong evidence suggesting a role for prenatal infections in SCZ development.35 However, given the presence of neurobiologic loci, like NOTCH4 and TRIM26, within the MHC, and the role of MHC class I in neuronal plasticity,11 we cannot rule out the possibility that the genetic associations found in the MHC may reflect shared pathobiology between SCZ and MS,11 in addition to immunological factors. Since myelin is affected in MS, the current findings are in line with previous work suggesting a role of abnormal myelination also in SCZ.36 Considered together, these findings strongly indicate the need for additional research to determine the MHC-related mechanisms underlying SCZ.
The three-way relationship between SCZ-MS-BD observed here and in our previous study14 demonstrates that overlap between SCZ and MS may be due to shared genetic risk loci mainly in the MHC, while overlap between BD and SCZ is present across almost the entire genome with the exception of the MHC. Indeed, removing the MHC-related SNPs did not affect the enrichment between BD and SCZ. Together with our current findings of no overlap between BD and MS, these results indicate that the MHC involvement could differentiate SCZ from BD susceptibility. The lack of pleiotropy between MS and BD indicates that the MHC is not associated with psychosis in general, but is more specifically related to SCZ. This is further supported by the lack of MS-conditional enrichment observed with autism spectrum disorder, major depressive disorder and ADHD (Supplementary Figure 7). Several lines of evidence support the notion of shared genes and phenotypes between SCZ and BD.3 Given that the SCZ and BD data sets entailed partly overlapping controls,7,13 the lack of findings in BD argues against unknown population stratification factors driving results for SCZ. Some yet unidentified population stratification related to immune genes within European populations37 could still have a role, but this would have also affected the results of the original GWAS.
The current findings of overlapping genetic variants in SCZ and MS show the feasibility of using a genetic epidemiology framework that leverages overlap in genetic signal from independent GWASs14,24,25 of brain phenotypes to improve statistical power for gene discovery. In the original SCZ GWAS sample, three loci were significant before replication in additional samples.7 By combining the original SCZ sample with the independent GWAS of MS, we identified significant pleiotropic signals in a total of 21 loci, only eleven of which had been reported in previous SCZ GWAS (Tables 1 and 2). This illustrates the increased power of our combined analytical approach. Note that the conditional FDR method protects against the possibility that the observed pleiotropic effects are driven by a strong signal in only one phenotype. It is likely that the new loci uncovered here would have been discovered if the number of subjects in the SCZ GWAS had been adequately large.38 However, we here show how summary statistics from independent samples provide a large advantage for gene discovery without the extra cost and resources needed to obtain new case–control samples. This has significant implications for GWAS, which have been criticized for not identifying enough of the genetic component of known heritable traits, and are increasingly seen as just a first step in more sophisticated analyses.39
Our findings also suggest that the etiology of complex brain disorders may involve a high number of pleiotropic variants, each with a small effect. SCZ appears to show an especially high degree of pleiotropy, with significant overlap with a number of different phenotypes.24 It is important to note, however, that this pleiotropy is not general, but is phenotype specific. The genetic pleiotropy observed between SCZ and MS is weaker than that between SCZ and BD.14 SCZ also shows varying degrees of pleiotropy with different CVD risk factors, with some showing strong effects (for example, triglyercides), others showing weaker effects (for example, blood pressure, waist-hip-ratio) and still others showing no significant enrichment (for example, type 2 diabetes24). With regard to SCZ pleiotropy with CVD risk, we showed that several of the SCZ loci detected here based on pleiotropy with MS were previously identified, leveraging pleiotropy between SCZ and CVD.24 This suggests the likely presence of genetic pleiotropy between MS and CVD risk factors that is worth further exploration.
To conclude, the conditional FDR approach identified 21 loci associated with SCZ conditioned on MS, and this enrichment was driven by the overlap in the MHC region. Imputation of the HLA alleles showed that the HLA alleles found to increase MS risk were associated with decreased SCZ risk. We found no overlap in the MHC between BD and MS, suggesting that MHC could be a key difference between SCZ and BD genetics. Our findings also indicate that leveraging existing GWASs for pleiotropic genes may represent an important avenue by which currently available resources can be optimized to identify more of the missing heritability of complex phenotypes.
References
Murray CJL . Health HSOP, World Health Organization Bank W. The globals burden of disease: A comprehensive assessment of mortality, injuries, and risk factors in 1990 and projected to 2020, 1st edn. Harvard School of Public Health: Cambridge MA, 1996.
Olesen J, Leonardi M . The burden of brain diseases in Europe. Eur J Neurol 2003; 10: 471–477.
Craddock N, Owen MJ . The beginning of the end for the Kraepelinian dichotomy. Br J Psychiatry 2005; 186: 364–366.
Editorial. A decade for psychiatric disorders. Nature 2010; 463: 9.
Arias I, Sorlozano A, Villegas E, de Dios Luna J, McKenney K, Cervilla J et al. Infectious agents associated with schizophrenia: a meta-analysis. Schizophr Res 2012; 136: 128–136.
Hope S, Melle I, Aukrust P, Steen NE, Birkenaes AB, Lorentzen S et al. Similar immune profile in bipolar disorder and schizophrenia: selective increase in soluble tumor necrosis factor receptor I and von Willebrand factor. Bipolar Disord 2009; 11: 726–734.
Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet 2011; 43: 969–976.
Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D et al. Common variants conferring risk of schizophrenia. Nature 2009; 460: 744–747.
Ripke S, O'Dushlaine C, Chambert K, Moran JL, Kähler AK, Akterin S et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet 2013; 45: 1150–1159.
Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
Shatz CJ . MHC class I: an unexpected role in neuronal plasticity. Neuron 2009; 64: 40–45.
Goldstein BI, Kemp DE, Soczynska JK, McIntyre RS . Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J Clin Psychiatry 2009; 70: 1078–1090.
Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 2011; 43: 977–983.
Andreassen OA, Thompson WK, Schork AJ, Ripke S, Mattingsdal M, Kelsoe JR et al. Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS Genet 2013; 9: e1003455.
Gourraud P-A, Harbo HF, Hauser SL, Baranzini SE . The genetics of multiple sclerosis: an up-to-date review. Immunol Rev 2012; 248: 87–103.
International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2, Sawcer S, Hellenthal G, Pirinen M, Spencer CCA, Patsopoulos NA, Moutsianas L et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011; 476: 214–219.
de Jager PL, Jia X, Wang J, de Bakker PIW, Ottoboni L, Aggarwal NT et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet 2009; 41: 776–782.
Gourraud P-A, Sdika M, Khankhanian P, Henry RG, Beheshtian A, Matthews PM et al. A genome-wide association study of brain lesion distribution in multiple sclerosis. Brain 2013; 136: 1012–1024.
Patsopoulos NA, Bayer Pharma MS . Genetics Working Group, Steering Committees of Studies Evaluating IFNβ-1b and a CCR1-Antagonist, ANZgene Consortium, GeneMSA, International Multiple Sclerosis Genetics Consortium et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol 2011; 70: 897–912.
Compston A, Coles A . Multiple sclerosis. Lancet 2008; 372: 1502–1517.
Takahashi N, Sakurai T, Davis KL, Buxbaum JD . Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog Neurobiol 2011; 93: 13–24.
Sivakumaran S, Agakov F, Theodoratou E, Prendergast JG, Zgaga L, Manolio T et al. Abundant pleiotropy in human complex diseases and traits. Am J Hum Genet 2011; 89: 607–618.
Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, van der Harst P et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet 2011; 43: 1131–1138.
Andreassen OA, Djurovic S, Thompson WK, Schork AJ, Kendler KS, O'Donovan MC et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet 2013; 92: 197–209.
Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet 2013; 45: 670–675.
Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium, Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013; 18: 497–511.
Cross-Disorder Group of the Psychiatric Genomics Consortium, Smoller JW, Craddock N, Kendler K, Lee PH, Neale BM et al. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013; 381: 1371–1379.
Neale BM, Medland SE, Ripke S, Asherson P, Franke B, Lesch K-P et al. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010; 49: 884–897.
Benjamini Y, Hochberg Y . Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Series B Stat Methodol 1995; 57: 289–300.
Schork AJ, Thompson WK, Pham P, Torkamani A, Roddey JC, Sullivan PF et al. All SNPs Are Not Created Equal: Genome-Wide Association Studies Reveal a Consistent Pattern of Enrichment among Functionally Annotated SNPs. PLoS Genet 2013; 9: e1003449.
Zheng X, Shen J, Cox C, Wakefield JC, Ehm MG, Nelson MR et al. HIBAG-HLA genotype imputation with attribute bagging. Pharmacogenomics J advance online publication, 28 May 2013; doi:10.1038/tpj.2013.18 (e-pub ahead of print).
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am J Hum Genet 2007; 81: 559–575.
Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe'er I et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009; 460: 753–757.
Hope S, Melle I, Aukrust P, Agartz I, Lorentzen S, Steen NE et al. Osteoprotegerin levels in patients with severe mental disorders. J Psychiatry Neurosci 2010; 35: 304–310.
Yolken RH, Torrey EF . Are some cases of psychosis caused by microbial agents? A review of the evidence. Mol Psychiatry 2008; 13: 470–479.
Karoutzou G, Emrich HM, Dietrich DE . The myelin-pathogenesis puzzle in schizophrenia: a literature review. Mol Psychiatry 2008; 13: 245–260.
Abi-Rached L, Jobin MJ, Kulkarni S, McWhinnie A, Dalva K, Gragert L et al. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 2011; 334: 89–94.
Sullivan PF, Daly MJ, O'Donovan M . Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet 2012; 13: 537–551.
Gershon ES, Alliey-Rodriguez N, Liu C . After GWAS: searching for genetic risk for schizophrenia and bipolar disorder. Am J Psychiatry 2011; 168: 253–256.
Acknowledgements
We thank all study participants for their contributions, the researchers involved in Psychiatric Genomics Consortium (PGC) Bipolar disorder and Schizophrenia Work Groups, the PGC ADHD, Autism, and MDD Groups, and the SURFsara Computer Cluster (www.surfsara.nl). The International Multiple Sclerosis Genetics Consortium and the Wellcome Trust Case Control Consortium2 is acknowledged for providing data from their MS genome-wide study, supported by the Wellcome Trust (project 085475/B/08/Z and 085475/Z/08/Z).
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Andreassen, O., Harbo, H., Wang, Y. et al. Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: differential involvement of immune-related gene loci. Mol Psychiatry 20, 207–214 (2015). https://doi.org/10.1038/mp.2013.195
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2013.195
Keywords
This article is cited by
-
Regulation of synaptic connectivity in schizophrenia spectrum by mutual neuron-microglia interaction
Communications Biology (2023)
-
fcfdr: an R package to leverage continuous and binary functional genomic data in GWAS
BMC Bioinformatics (2022)
-
Large-scale genomic analyses reveal insights into pleiotropy across circulatory system diseases and nervous system disorders
Nature Communications (2022)
-
Characterising the shared genetic determinants of bipolar disorder, schizophrenia and risk-taking
Translational Psychiatry (2021)
-
Shared genetic etiology underlying Alzheimer’s disease and major depressive disorder
Translational Psychiatry (2020)
