
Abstract
The core features of schizophrenia include deficits in cognitive processes mediated by the circuitry of the dorsolateral prefrontal cortex (DLPFC). These deficits are associated with a range of molecular and morphological alterations in the DLPFC, each of which could be a cause, consequence, or compensation in relation to other changes, and thus reflect the neuroplasticity of the brain in response to the underlying disease process. In this review, we consider disturbances in excitatory, inhibitory, and modulatory connections of DLPFC circuitry from the perspective of disease- and development-related neuroplasticity and discuss their implications for the identification of novel therapeutic targets.
Similar content being viewed by others
INTRODUCTION
In a broad sense of the term, neuroplasticity refers to the capacity of the brain to change the molecular and structural features that dictate its function. In the context of a disease process such as schizophrenia, these changes, as observed directly in postmortem tissue or reflected in biomarkers detectable by neuroimaging, electroencephalography or other noninvasive assessments of brain structure and function, could represent any of the following four ‘Cs’: (1) Cause, an upstream factor related to the disease pathogenesis; (2) Consequence, a deleterious effect of a cause; (3) Compensation, a response to either cause or consequence that helps restore homeostasis; or (4) Confound, a product of factors frequently associated with, but not a part of, the disease process, or an artifact of the approach used to obtain the measure of interest. Thus, understanding neuroplasticity (as defined above) in schizophrenia requires the identification of the brain alterations that are reliably associated with the clinical syndrome that we term schizophrenia, and then the determination of the ‘C’ category to which each alteration belongs.
This process is, of course, complicated by the apparent heterogeneity associated with the diagnosis of schizophrenia. From the classic view of a disease process, the etiology of schizophrenia unleashes pathogenetic mechanisms that produce a pathological entity, a conserved set of molecular and cellular disturbances in the brain. This pathological entity so alters the brain's normal function that the resulting pathophysiology gives rise to the emergent properties recognized as the clinical features of the illness (Lewis et al, 2005). Etiologically, most cases of schizophrenia are thought to be the consequence of a complex interplay of genetic liabilities and environmental risk factors that alter the developmental trajectories of neural circuits (Harrison and Weinberger, 2005; Lewis and Levitt, 2002). At the other end of the disease process, the broad range of clinical features found in individuals who meet diagnostic criteria for schizophrenia suggests that multiple brain systems are affected in this disorder. Thus, a critical challenge in exploring neuroplasticity in schizophrenia is to understand this heterogeneity from the perspectives of diversity (the existence of different disease entities within the population meeting diagnostic criteria) and variability (variance of particular parameters within a disease entity).
One approach to addressing this problem is to focus on a well-defined clinical component of the illness. In this regard, it has been argued that among the clinical manifestations of schizophrenia, the deficits in cognitive abilities are the core features of the illness (Elvevag and Goldberg, 2000). For example, a characteristic pattern of cognitive deficits occurs with high frequency in individuals with schizophrenia, is relatively stable over the course of the illness, is independent of the psychotic symptoms of the disorder, and is present in a milder form in individuals at genetic risk who do not become clinically ill (Gold, 2004). Furthermore, the degree of cognitive impairment is the strongest determinant of functional outcome for individuals with schizophrenia (Green, 1996).
Of the different domains of cognitive dysfunction in schizophrenia, working memory, the ability to transiently maintain and manipulate a limited amount of information to guide thought or behavior, has been studied extensively. Working memory involves several component processes including storage buffers for different types of information (eg visuo-spatial scratch pad (short-term storage of visual information) and phonological loop (articulatory rehearsal and phonological coding of verbal information) and a central executive component that controls the manipulation of information within the storage buffers (Baddeley, 1992). In subjects with schizophrenia, tasks that depend primarily on the phonological loop show relatively little impairment. In addition, activation of the brain regions (eg ventral lateral prefrontal cortex and posterior parietal cortex) that mediate the components of the phonological loop appear to be intact (Barch, 2006). In contrast, central executive function, especially the manipulation of transiently stored information, is clearly disturbed in subjects with schizophrenia (Cannon et al, 2005; Tan et al, 2005), and this dysfunction is accompanied by altered activation of the dorsolateral prefrontal cortex (DLPFC) (Box 1), a brain region known to be associated with executive function (Callicott et al, 2003). The altered activation of the DLPFC under such conditions might be specific to the disease process of schizophrenia because these disturbances are present in medication-naive individuals with schizophrenia, but not in subjects with other psychotic disorders or major depression (Barch et al, 2003; MacDonald et al, 2005).
Consequently, in this paper, we examine the alterations in excitatory, inhibitory, and modulatory components of DLPFC circuitry in schizophrenia that might contribute to the impairments in working memory. Each component is examined in relation to the four ‘Cs’ and considered from the perspectives of disease and developmental neuroplasticity. It is important to note that while we focus on the DLPFC as a prototypic cortical region affected in the illness, the alterations described below are likely to be present in multiple other cortical regions as well.
NEUROPLASTICITY OF EXCITATORY CORTICAL CONNECTIONS IN SCHIZOPHRENIA
Evidence of Altered Excitatory Connections in the DLPFC in Schizophrenia
The disease process of schizophrenia appears to involve deficient glutamate-mediated excitatory neurotransmission through the NMDA receptor (Moghaddam, 2003; Olney and Farber, 1995). NMDA receptor antagonists such as phencyclidine (PCP) or ketamine increase both positive and negative symptoms in patients with schizophrenia (Coyle, 2004), and the administration of subanesthetic doses of ketamine to healthy individuals produces thought disorder and other features similar to those seen in schizophrenia (Adler et al, 1999; Krystal et al, 1994). In addition, systemic administration of NMDA receptor antagonists disrupts working memory in rats (Verma and Moghaddam, 1996), and application of an NMDA receptor antagonist to the DLPFC (but not to the primary visual cortex) impairs working memory performance in monkeys (Dudkin et al, 2001). Furthermore, computational modeling studies of neuronal network activity in the DLPFC suggest that an NMDA receptor activation supports slow reverberating excitation (Durstewitz and Seamans, 2006; Wang, 2006). This pattern of excitation is critical for the sustained firing of DLPFC neurons that is considered to be the cellular basis of working memory (Goldman-Rakic, 1995).
Hypoactivation of only NMDA receptors in schizophrenia is unlikely to result from deficits in glutamate release because, in the adult monkey DLPFC, both AMPA and NMDA receptors mediate fast synaptic transmission at glutamate synapses (Gonzalez-Burgos et al, 2007b); thus, decreased glutamate release would also result in deficits in AMPA receptor-mediated transmission. Consequently, selective NMDA receptor hypofunction would appear more likely to result from postsynaptic deficits in NMDA receptor-mediated signaling. However, although postmortem studies have reported alterations in measures of glutamate receptor binding, transcription and subunit protein expression in several brain regions in subjects with schizophrenia (Konradi and Heckers, 2003), such findings for mRNA and protein levels of NMDA receptor subunits in the DLPFC have been limited in magnitude and not always replicated (Akbarian et al, 1996; Dracheva et al, 2001; Kristiansen et al, 2006; Mirnics et al, 2000). Indeed, as noted elsewhere (Kristiansen et al, 2007), the modest and inconsistent findings in postmortem studies of NMDA receptor transcripts and protein expression levels in schizophrenia suggest that other components of NMDA receptor signaling might be affected in the illness (Kristiansen et al, 2007). It is possible, however, that NMDA hypoactivity in schizophrenia is associated with decreased levels of synaptic NMDA receptor proteins in particular classes of inputs that are difficult to detect.
Anatomical studies do support the presence of input-specific alterations of excitatory connections in the DLPFC in schizophrenia. In the DLPFC, pyramidal neurons are the principal source of glutamate neurotransmission, as well as the targets of the majority of glutamate-containing axon terminals (White, 1989). Although the number of these neurons does not appear to be altered in schizophrenia (Akbarian et al, 1995; Thune et al, 2001), neuronal density in the DLPFC and certain other cortical regions, such as the primary visual cortex, has been reported to be increased in schizophrenia (Selemon et al, 1995). (However, more recent studies have failed to support this finding, at least in the visual cortex; Dorph-Petersen et al, 2007.) Increased cell packing density has been interpreted as evidence of a reduction in the amount of cortical neuropil, the axon terminals, dendritic spines, and glia processes that occupy the space between neurons (Selemon and Goldman-Rakic, 1999). Consistent with this interpretation, decreased synaptophysin protein, a marker of axon terminals, has been reported to be decreased in the DLPFC of subjects with schizophrenia (Glantz and Lewis, 1997; Karson et al, 1999; Perrone-Bizzozero et al, 1996). Furthermore, gene expression profiling studies have found reduced tissue levels of gene transcripts that encode proteins involved in the presynaptic regulation of neurotransmission (Mirnics et al, 2000).
Dendritic spines are the principal targets of excitatory synapses to pyramidal neurons. Approximately 75% of the dendritic spines present in the somatosensory cortex of young adult mice are numerically stable over adulthood (Zuo et al, 2005), but they are subject to a number of neuroplastic changes, such as a loss of their presynaptic excitatory input. In schizophrenia, dendritic spine density in pyramidal neurons has been reported to be lower in both the DLPFC and other cortical regions by multiple research groups (Black et al, 2004; Broadbelt et al, 2002; Garey et al, 1998; Glantz and Lewis, 2000; Kalus et al, 2000). These findings have raised a series of questions regarding the neuroplastic nature of the apparent changes in spine density in schizophrenia.
Is the Lower Spine Density in Schizophrenia Specific to Certain Subpopulations of DLPFC Pyramidal Neurons?
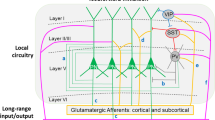
Pyramidal neurons can be divided into subgroups based on the brain region targeted by their principal axonal projection and the sources of their excitatory inputs; both of these characteristics are associated with the laminar location of pyramidal cell bodies (Box 1). For example, many pyramidal cells in layers 2 and 3 send axonal projections to other cortical regions, pyramidal neurons in layer 5 tend to project to the striatum and other subcortical structures, and pyramidal neurons in layer 6 furnish projections primarily to the thalamus (Jones, 1984). Studies of basilar dendritic spine density on Golgi-impregnated pyramidal neurons in each cortical layer of the DLPFC in the same cohort of subjects found a significant effect of diagnosis on spine density only for pyramidal neurons in deep layer 3 (Figure 1; Glantz and Lewis, 2000; Kolluri et al, 2005). In subjects with schizophrenia, spine density on these neurons was significantly decreased by 23 and 16%, respectively, compared to normal and psychiatrically ill comparison subjects. In contrast, spine density on pyramidal neurons in superficial layer 3 was reduced to a lesser degree, whereas the density of those on the basilar dendrites of pyramidal neurons in layer 5 or 6 did not differ across subject groups.

Pyramidal neuron dendritic spines in the human dorsolateral prefrontal cortex (DLPFC). (a) Schematic diagram illustrating the dendritic tree and dendritic spines on a prototypic pyramidal neuron. (b) Electron micrograph showing a dendrite (D) with two spines (S). Each spine receives an asymmetric (presumably excitatory) synapse from an axon terminal (at). (c) Golgi-impregnated basilar dendrites and spines on deep layer 3 pyramidal neurons from a normal comparison (top) and two subjects with schizophrenia (bottom). Note the reduced density of spines in the subjects with schizophrenia in these extreme examples. (d) Scatter plot demonstrating the lower density of spines on the basilar dendrites of deep layer 3 pyramidal neurons in the DLPFC of subjects with schizophrenia relative to both normal and psychiatrically ill comparison subjects. (e) Laminar-specificity of the spine density differences in the same subjects. (c–e) Adapted from Glantz and Lewis (2000); Kolluri et al (2005).
The functional integrity of the pyramidal neurons with lower dendritic spine densities may be reflected in changes in their somal volume. For example, shifts in somal size may indicate disturbances in neuronal connectivity, given that somal size has been shown to be correlated with measures of a neuron's dendritic tree (Hayes and Lewis, 1996; Jacobs et al, 1997) and axonal arbor (Gilbert and Kelly, 1975; Lund et al, 1975). Indeed, the mean cross-sectional somal area of the Golgi-impregnated, deep layer 3 pyramidal neurons was 9.1% smaller in the subjects with schizophrenia relative to normal control subjects, although this difference did not achieve statistical significance (Glantz and Lewis, 2000). Consistent with this observation, the mean somal volume of Nissl-stained pyramidal neurons in DLPFC deep layer 3 was 9.2% smaller in a different cohort of subjects with schizophrenia, relative to matched normal comparison subjects, a decrease that was not explained by either antipsychotic medication history or duration of illness (Pierri et al, 2001). Similarly, in another study, the mean somal size of all layer 3 neurons in DLPFC area 9 was smaller in subjects with schizophrenia, and was accompanied by a decrease in the density of the largest neurons in deep layer 3, without a change in somal volume in layer 5 (Rajkowska et al, 1998). Furthermore, in both primary and association auditory cortices, somal volumes of deep layer 3, but not of layer 5, pyramidal neurons were smaller in schizophrenia (Sweet et al, 2003, 2004). Together, these findings suggest that in schizophrenia, (1) basilar dendritic spine density is lower and somal volume is smaller in deep layer 3 pyramidal neurons, (2) these alterations are specific to or at least most prominent in deep layer 3, (3) this pattern of alterations is not restricted to the DLPFC, and (4) these differences reflect the underlying disease process and not confounding factors. However, these findings do not reveal the biochemical or connectional identity of the affected pyramidal neurons.
Even within the same cortical layer, different subpopulations of pyramidal neurons exhibit quantitative differences in dendritic morphology and qualitative differences in the gene products that they express, and these subpopulations of pyramidal neurons tend to differ in the targets of their principal axon projections. For example, pyramidal neurons in the supragranular layers of the monkey DLPFC with axons that project callosally have larger dendritic arbors and a greater density of dendritic spines than do neighboring pyramidal cells that furnish axons to the adjacent regions of the ipsilateral DLPFC (Duan et al, 2002; Soloway et al, 2002). Pyramidal neurons that furnish axonal projections to distant cortical regions also tend to have larger cell bodies and to express high levels of non-phosphorylated epitopes of neurofilament proteins (NNFPs) compared to pyramidal cells in the same location that provide shorter corticocortical projections (Hof et al, 1996). Attempts to identify the affected pyramidal neurons based on such markers have not yet yielded fruit. For example, the somal volumes of NNFP-containing layer 3 neurons were reported to be unaltered in schizophrenia (Law and Harrison, 2003; Pierri et al, 2003), but these studies appear to have been subject to a methodological confound that resulted in an overestimation of somal volumes in schizophrenia (Maldonado-Aviles et al, 2006). Furthermore, the ability to distinguish other subpopulations of pyramidal neurons based on their molecular phenotype still awaits the types of gene expression profiling studies that have been successfully utilized for characterizing subclasses of cortical interneurons (Sugino et al, 2006).
Are the Reductions in Spine Density Associated with a Deficit in a Specific Source of Excitatory Inputs?
Excitatory projections from the mediodorsal thalamus, the principal source of thalamic inputs to the DLPFC (Giguere and Goldman-Rakic, 1988), synapse primarily on dendritic spines (Melchitzky et al, 1999). These axons densely arborize in DLPFC layers deep 3 and 4, but do not innervate the deep cortical layers (Erickson and Lewis, 2004). Since the laminar distributions of these afferents matches that of the basilar dendrites of DLPFC deep layer 3 pyramidal neurons, a reduction in the number or activity of these afferents could contribute to the observed decrement in spine density in schizophrenia. Initial reports indicated that the total number of neurons in this nucleus was lower in schizophrenia (Byne et al, 2002; Pakkenberg, 1990; Popken et al, 2000; Young et al, 2000), suggesting that a reduced number of these afferents might contribute to lower spine density in the DLPFC in schizophrenia. However, more recent studies, including some from the same research groups that reported the initial positive findings, have failed to find a decrement in thalamic neuron number (Cullen et al, 2003; Dorph-Petersen et al, 2004; Kreczmanski et al, 2007; Young et al, 2004). Some neuroimaging studies have reported smaller thalamic volumes, altered shape and decreased activity (Manoach et al, 1999) in subjects with schizophrenia, including studies in first-episode subjects in whom the potential confounds of medication effects and illness chronicity are mitigated (Gilbert et al, 2001). Thus, even in the face of a normal number of thalamic neurons, these observations might suggest that the nature, if not the number, of thalamic projections is altered in schizophrenia. However, a reduction in thalamic inputs to the DLPFC cannot completely account for the decrease in dendritic spine density since thalamocortical terminals appear to comprise a small proportion (<10%) of the total excitatory inputs to the targeted cortical neurons in animals (Guillery and Sherman, 2002). If this holds true in the human DLPFC, then (in the absence of some synergistic factor—see below) even a total absence of thalamocortical afferents would not be sufficient to account for the apparent magnitude of the reduction in basilar dendritic spine density on deep layer 3 pyramidal cells observed in schizophrenia.
Two other major sources provide excitatory inputs to deep layer 3 pyramidal neurons in the primate DLPFC, although these inputs are less laminarly restricted than those from the mediodorsal thalamus. First, the axons of pyramidal neurons in layers 2 and 3, which project to other cortical regions, also give rise to both local (within 300 μm of the cell body) and long-range axon collaterals that travel through the gray matter for up to several millimeters before arborizing in discrete stripe-like clusters in the same cortical region (Levitt et al, 1993; Pucak et al, 1996). Both the extrinsic and long-range intrinsic collaterals of these neurons target almost exclusively the dendritic spines of other pyramidal cells, whereas the synaptic targets of the local axon collaterals are equally divided between dendritic spines and the dendritic shafts of the parvalbumin-containing class of GABA neurons (Melchitzky et al, 1998, 2001; Melchitzky and Lewis, 2003). Second, associational or callosal projections from other cortical regions also terminate in these layers (Barbas, 1992; Pucak et al, 1996). Thus, the smaller decrease in spine density on superficial layer 3 (relative to deep layer 3) pyramidal cells raises the possibility that abnormalities in thalamocortical afferents to deep layer 3 have an additive effect to a disturbance in the cortical axon terminals that are distributed across layer 3. However, even if these interpretations are correct, they do not reveal the direction of the pathogenetic mechanisms. For example, the reduced number of excitatory inputs to DLPFC layer 3 pyramidal cells might not be the consequence of a more primary disturbance in the source of these afferents, but an abnormality intrinsic to these pyramidal cells could render them unable to support a normal complement of excitatory inputs.
What Molecular Mechanisms Mediate the Lower Spine Density in Schizophrenia?
Attempts to explore the direction of these pathogenetic mechanisms have looked at both extracellular and intracellular molecular mediators of spine number in schizophrenia. One potential extracellular mediator is the secreted neurotrophin, brain-derived neurotrophic factor (BDNF). BDNF stimulates the growth of dendrites and increases the spine density of cortical pyramidal neurons in organotypic brain slices in a lamina-specific fashion (McAllister et al, 1995, 1996). The expression of BDNF mRNA is lower in the DLPFC of subjects with schizophrenia (Hashimoto et al, 2005; Weickert et al, 2003), and BDNF mRNA levels and spine density on the basilar dendrites of deep layer 3 pyramidal neurons were positively correlated (r=0.91; p=0.004) in the same subjects with schizophrenia (Hill et al, 2005). However, large (∼80%) genetically engineered reductions in BDNF expression, induced either prenatally or in early adulthood, were not sufficient to alter basilar dendritic morphology in the prefrontal cortex of adult mice (Hill et al, 2005). Similarly, in heterozygous bdnf knockout mice, a 50% reduction in BDNF protein levels did not affect the amount of cortical neuropil (Genoud et al, 2004), of which excitatory axon terminals and dendritic spines are a major component. Therefore, these findings suggest that reduced levels of BDNF mRNA expression in subjects with schizophrenia are not sufficient to account for the observed deficit in spine density. However, it must be kept in mind that potential compensatory mechanisms, such as increased expression of the neurotrophin NT-4 or of the receptor for BDNF, tyrosine kinase B (trkB), might occur in bdnf knockout mice but not in schizophrenia. For example, in subjects with schizophrenia, reduced BDNF levels are accompanied by a significant decrease in trkB mRNA levels (Hashimoto et al, 2005; Weickert et al, 2005). Thus, reduced levels of both BDNF and trkB may be required to produce the decrease in spine density observed in schizophrenia. Alternatively, the murine models may fail to reproduce a cell type-specific decrement in BDNF mRNA expression in schizophrenia that is required for alterations in spine density.
An increasing number of intracellular signaling pathways have been shown to regulate the actin cytoskeleton that is critical for spine structure (Calabrese et al, 2006). In neurons, different members of the RhoGTPase family regulate specific aspects of dendritic morphology and spine number (Luo, 2000; Negishi and Katoh, 2005). For example, the RhoGTPase Cdc42 (cell division cycle 42), which regulates the outgrowth of the actin cytoskeleton, is involved in the formation of thin protrusions (filopodia), which may develop into mature spines. In neuronal cultures, Cdc42 is essential for the establishment of the full complement of dendritic spines (Ramakers, 2002; Scott et al, 2003). Rac1 (Ras-related C3 botulinum toxin substrate 1) helps to stabilize the actin cytoskeleton, and thus appears to contribute to the outgrowth of spine-like structures (Nobes and Hall, 1995; Ramakers, 2002) and to be a critical regulator of dendritic spine density (Nakayama et al, 2000). In contrast, RhoA (Ras homologous member A) regulates the destabilization of the actin cytoskeleton, such that the activation of RhoA results in a reduction in the density of dendritic spines (Nakayama et al, 2000; Nobes and Hall, 1995). Other proteins that are selectively localized to spines also play roles in spine structure. Duo, the human ortholog of the murine Kalirin-7, is a GDP–GTP exchange factor that activates Rac1 (Penzes et al, 2000) and is essential for spine maintenance and for coordinating the apposition of presynaptic terminals to the sites of postsynaptic densities (Ma et al, 2003; Penzes et al, 2001). Drebrin, an actin-binding protein (Hayashi et al, 1996), regulates the maturation of filopodia into mature spines by promoting the formation of the postsynaptic density and appears to be involved in spine morphogenesis by promoting the conversion of filopodia into mature spines (Takahashi et al, 2003).
Studies of the transcripts for these five proteins in the DLPFC of subjects with schizophrenia revealed that only the mRNAs for Cdc42 and Duo showed significantly decreased expression levels that were (1) restricted to the cortical gray matter; (2) significantly correlated with spine density in the same subjects; and (3) not due to chronic treatment with antipsychotic medications or other potential confounds (Hill et al, 2006). Because Cdc42 regulates the polymerization of the actin cytoskeleton into filopodia (Nobes and Hall, 1995), many of which are precursors for mature spines (Dailey and Smith, 1996), reduced levels of Cdc42 might impair the ability of pyramidal neurons to generate new filopodia in the adult neocortex. Furthermore, the formation of spines is closely tied to the generation of stable constructs of the actin cytoskeleton (Nobes and Hall, 1995). This stability is regulated by Rac1, whose upstream regulator is Duo (Carlisle and Kennedy, 2005). Therefore, a decrease in Duo mRNA expression would result in reduced activation of Rac1, and consequently impaired stability of dendritic spines. In combination, a decrease in the expression of both Cdc42 and Duo mRNAs would be expected to result in a reduction in the formation of new spines and impaired maintenance of existing, mature spines.
However, expression levels of both Cdc42 and Duo mRNAs were comparable in both deep layer 3 and 6 in the subjects with schizophrenia. Although this observation suggests that the decreased expression of Cdc42 and Duo mRNAs in schizophrenia is not a consequence of a reduced number of pyramidal neuron dendritic spines, since the expression of these mRNAs was similarly reduced in pyramidal neurons with or without a reduction in spine density, it also indicates that reduced expression of Cdc42 and Duo mRNAs is not sufficient to cause reduced spine density. Because excitatory synaptic activity (via NMDA receptors) can stimulate the formation of new spines, in addition to changing the shape of existing spines (Carlisle and Kennedy, 2005; Jourdain et al, 2003), a laminar-specific reduction in excitatory inputs, such as those discussed above, might interact with alterations in the molecular cascades involving Cdc42 and Duo to produce a reduction in spine density in schizophrenia. Although speculative, this interpretation suggests the testable hypothesis that spine density is decreased on the portions of the apical dendrites of pyramidal neurons in layer 5 or 6 that pass through layer deep 3, while being unaltered on the basilar dendrites of the same neurons (Figure 2).

Schematic diagram summarizing the idea that reduced dendritic spine density in schizophrenia might require alterations both in regulators of the actin cytoskeleton that are intrinsic to pyramidal neurons and in the extrinsic excitatory inputs to pyramidal neurons. In this example, reduced expression of Cdc42 and Duo mRNAs and reduced inputs from the mediodorsal (MD) nucleus of the thalamus or the intrinsic axon collaterals of layer 3 pyramidal neurons are hypothesized to produce the apparent laminar-specific reduction in dendritic spine density on the basilar dendrites of deep layer 3 pyramidal neurons in subjects with schizophrenia. As illustrated in the figure, the apical dendrites of pyramidal neurons in layer 6 that extend through the thalamic termination zone are predicted to show a reduction in spine density that is similar to that observed on the basilar dendrites of pyramidal neurons in deep layer 3. In contrast to the restricted termination of excitatory inputs from the thalamus and intrinsic axon collaterals to specific cortical layers, those originating in other cortical association regions tend to terminate across all cortical layers. Thus, changes in these inputs would be expected to produce alterations in spine density across all cortical layers, unless critical regulators of the actin cytoskeleton that determine spine development, stability or retraction are selectively altered in a subset of pyramidal neurons. Adapted from Hill et al (2006).
It must also be kept in mind that other molecular cascades contribute to the regulation of the actin cytoskeleton (Carlisle and Kennedy, 2005), and their potential role in the reduced density of dendritic spines in schizophrenia remains to be determined. For example, inhibitors of spine formation, such as Nogo, which has been reported to be increased in the frontal cortex in schizophrenia (Novak and Tallerico, 2006), could contribute to the observed reductions in spine density. In addition, it could be that molecules selectively expressed by layer 3 pyramidal neurons (Arion et al, 2007) provide the essential factor that determines the apparent heightened vulnerability of these neurons in schizophrenia.
Does Developmental Plasticity Contribute to Dendritic Spine Alterations in Schizophrenia?
Dendritic spine density on DLPFC layer 3 pyramidal neurons undergoes a substantial decline during adolescence in primates (Anderson et al, 1995). Consistent with the findings that dendritic spines are the main site of excitatory synaptic input onto pyramidal cells and that all mature dendritic spines contain an excitatory synapse (Arellano et al, 2007), the number of asymmetric (presumably excitatory) synapses declines in a similar age-related fashion in both monkey and human DLPFC (Bourgeois et al, 1994; Huttenlocher and Dabholkar, 1997). In humans, this synaptic pruning is thought to underlie the decrease in cortical gray matter thickness that occurs during adolescence (Giedd, 1999; Gogtay et al, 2004). Interestingly, the late developmental refinements in excitatory connectivity are more marked in layer 3 than in the deeper cortical layers (Bourgeois et al, 1994), suggesting that they may be associated with the apparent lamina-specific alterations in spine density in schizophrenia. The observation of alterations in the expression of certain synaptic proteins in schizophrenia suggested the possibility that the exuberant synapses present before adolescence somehow compensated for a dysfunction in excitatory transmission in individuals with schizophrenia (Mirnics et al, 2001). Alternatively, such alterations in synaptic protein expression might disturb the mechanisms of adolescence-related synapse elimination leading, for instance, to excessive synapse pruning and decreased spine number in the illness (Feinberg, 1982; Keshavan et al, 1994).
Understanding how reductions in excitatory synaptic density during adolescence could contribute to disease-related changes in DLPFC function depends, in part, on knowledge of the functional properties of the synapses that are pruned. Current views on the mechanisms by which synapses are eliminated during early brain development suggest that the pruned synapses are functionally immature. Immature glutamate synapses are relatively weak and their maturation involves an activity-dependent increase in strength. Such activity-dependent strengthening might underlie synapse stabilization, and thus mark for elimination of the immature synapses that are not strengthened (Katz and Shatz, 1996; Le Be and Markram, 2006; Waites et al, 2005). Furthermore, spine morphology correlates with synaptic strength, that is, spines with smaller spine heads have a smaller postsynaptic density, fewer AMPA receptors and a weaker response to glutamate (Matsuzaki et al, 2001; Matsuzaki, 2007). Moreover, patterns of activity consistent with those that induce long-term potentiation of synaptic strength may increase spine head size concomitantly with the synapse-specific increase in strength (Matsuzaki, 2007). Conversely, activity patterns that produce long-term depression may lead to a reduction in synaptic strength together with spine head shrinkage and eventually spine disappearance (Matsuzaki, 2007; Zhou et al, 2004). Activity-dependent changes in dendritic spine shape and size, as well as spine formation and retraction, are dependent on regulation of actin dynamics and clathrin-dependent membrane endocytosis (Blanpied and Ehlers, 2004; Tada and Sheng, 2006).
Determining if the synapses that are eliminated during adolescence in the primate DLPFC are functionally immature is essential for understanding how synaptic pruning might inform neurodevelopmental models of schizophrenia. For example, if the eliminated synapses are functionally immature, then they may not be able to provide the hypothesized compensation for synaptic dysfunction prior to adolescence in individuals with schizophrenia (Mirnics et al, 2001). This lack of compensatory capacity is due to the facts that neurotransmission at immature synapses has (1) very low AMPA receptor contribution, rendering these synapses silent at the resting membrane potential; and (2) relatively high probability of glutamate release, such that these synapses are not able to be repetitively activated without quickly exhausting glutamate vesicle pools (Schneggenburger et al, 2002; Zucker and Regehr, 2002).
In a living slice preparation of the monkey DLPFC (Gonzalez-Burgos et al, 2007b), excitatory inputs to layer 3 pyramidal neurons in 3-month-old monkeys (early postnatal), had immature functional properties, including a higher probability of glutamate release, lower AMPA/NMDA receptor ratio and a longer duration of NMDA receptor-mediated excitatory synaptic currents. In contrast, the excitatory synaptic inputs to layer 3 pyramidal neurons from 15-month-old monkeys (pre-adolescence and pre-pruning) had mature functional properties that were similar to those observed in neurons from 42-month-old (post-adolescence and post-pruning) and 84-month-old (adult) animals. Therefore, the contribution of functionally immature synapses decreases significantly before synapse elimination begins, and remains essentially constant thereafter. These data suggest that the substantial remodeling of excitatory connectivity of the primate DLPFC during adolescence primarily involves the elimination of mature synapses, and that some other factor, such as the neuronal source of input, somehow tags mature synapses for pruning (Woo et al, 1997b).
Recent imaging studies of the dynamics of neocortical dendritic spines and synaptic boutons in vivo suggest that synaptogenesis is an active process throughout life (Holtmaat et al, 2006; Knott et al, 2006; Stettler et al, 2006). Thus, synapse density at a given stage of development depends on the rates of active synapse formation and elimination (reviewed in Alvarez and Sabatini, 2007). For instance, in the adult monkey visual cortex in vivo, about 7% of the synapses are lost per week, but the rate of synapse formation is very similar, resulting in no net change in synapse density (Stettler et al, 2006). If these findings are true for the human DLPFC, then lower synapse density in schizophrenia could result from decreased synapse formation or increased synapse elimination at any developmental stage, including adulthood.
NEUROPLASTICITY OF INHIBITORY CORTICAL CONNECTIONS IN SCHIZOPHRENIA
Evidence of Altered Inhibitory Connections in the DLPFC in Schizophrenia
Initial postmortem studies found evidence of decreased glutamic acid decarboxylase (GAD) activity (Bird et al, 1979), decreased GABA reuptake (Simpson et al, 1989) and increased binding to GABAA receptors (Benes et al, 1996; Hanada et al, 1987) in the neocortex of subjects with schizophrenia. Studies conducted over the past decade, using DNA microarray, real-time quantitative PCR or in situ hybridization, have consistently found reduced levels of the transcript for the 67-kDa isoform of glutamic acid decarboxylase (GAD67), the principal synthesizing enzyme for GABA, in the DLPFC of subjects with schizophrenia (Akbarian et al, 1995; Guidotti et al, 2000; Hashimoto et al, 2005, 2007a; Mirnics et al, 2000; Straub et al, 2007; Vawter et al, 2002; Volk et al, 2000). Similar findings have also been reported in other neocortical regions (Akbarian and Huang, 2006; Woo et al, 2004). Indeed, an analysis of all postmortem studies of schizophrenia conducted in specimens from the Stanley Neuropathology Consortium revealed that three genes expressed in GABA neurons (reelin, parvalbumin, and GAD67) had the most abnormal transcript and protein levels in schizophrenia (Torrey et al, 2005). At the cellular level, the expression of GAD67 mRNA was not detectable in ∼25–35% of GABA neurons in layers 1–5 of the DLPFC, but the remaining GABA neurons exhibited normal levels of GAD67 mRNA (Akbarian et al, 1995; Volk et al, 2000). Furthermore, levels of the mRNA for the GABA membrane transporter (GAT1), a protein responsible for reuptake of released GABA into nerve terminals, was also decreased (Ohnuma et al, 1999), and this decrease was restricted to a similar minority of GABA neurons (Volk et al, 2001). These findings suggest that both the synthesis and reuptake of GABA are lower in a subset of DLPFC neurons in schizophrenia.
Which Subclasses of DLPFC GABA Neurons are Affected in Schizophrenia?
As in other cortical regions, the inhibitory GABA neurons in the primate DLPFC appear to form subclasses that can be distinguished on the basis of a number of molecular, electrophysiological, and anatomical properties. For example, as illustrated in Box 2, the Ca2+-binding proteins, parvalbumin (PV) and calretinin (CR), and the neuropeptide somatostatin (SST) are, with a few exceptions, expressed in separate populations of cortical GABA neurons (Condé et al, 1994; DeFelipe, 1997; Gabbott and Bacon, 1996). These subtypes tend to exhibit different membrane-firing properties (Kawaguchi and Kubota, 1993; Krimer et al, 2005; Zaitsev et al, 2005) and to have axons with different arborization patterns and synaptic targets (DeFelipe, 1997). For example, the axon terminals of PV-containing chandelier and basket neurons principally target the axon initial segments and cell body/proximal dendrites, respectively, of pyramidal neurons (Melchitzky et al, 1999; Williams et al, 1992), the SST-containing Martinotti cells innervate the distal dendrites of pyramidal neurons (DeLima and Morrison, 1989; Kawaguchi and Kubota, 1996; Ma et al, 2006), and the CR-containing double-bouquet cells tend to synapse on the dendrites of other GABA cells (Melchitzky et al, 2005).
The affected GABA neurons in schizophrenia include the PV-positive neurons, which comprise ∼25% of GABA neurons in the primate DLPFC. In individuals with schizophrenia the expression level of PV mRNA is reduced, although the number of PV neurons appears to be unchanged (Woo et al, 1997a); in addition, approximately half of PV mRNA-containing neurons lack detectable levels of GAD67 mRNA (Hashimoto et al, 2003). In contrast, the ∼50% of GABA neurons that express CR appear to be unaffected (Hashimoto et al, 2003).
In the DLPFC of subjects with schizophrenia, GAT1 immunoreactivity is selectively reduced in the characteristic axon terminals (cartridges) of the chandelier class of PV-containing neurons (Woo et al, 1998). In the postsynaptic targets of these axon cartridges, the axon initial segments of pyramidal neurons, immunoreactivity for the GABAA receptor α2-subunit (which is present in most GABAA receptors in this location; Nusser et al, 1996) is markedly increased in schizophrenia (Volk et al, 2002). These changes appear to be specific to the disease process of schizophrenia because they are not found in subjects with other psychiatric disorders or in monkeys exposed chronically to antipsychotic medications (Hashimoto et al, 2003; Volk et al, 2000, 2001, 2002).
Several lines of evidence suggest that the reductions in presynaptic GABA markers (GAT1 and PV) and increased postsynaptic GABAA receptors are compensatory responses to a deficit in GABA release from chandelier neurons. For example, PV is a slow Ca2+ buffer that does not affect the amplitude, but accelerates the decay, of Ca2+ transients in GABA nerve terminals (Collin et al, 2005; Muller et al, 2007). Thus, PV decreases the residual Ca2+ levels that normally accumulate in nerve terminals and facilitate GABA release during repetitive firing (Collin et al, 2005). Studies in PV-deficient mice have demonstrated that a decrease in PV increases residual Ca2+ and favors synaptic facilitation (Collin et al, 2005; Vreugdenhil et al, 2003). Furthermore, the enhanced facilitation of GABA release from fast-spiking neurons with reductions in PV is associated with increased power of γ oscillations (Vreugdenhil et al, 2003) (see below). Similarly, the blockade of GABA reuptake via GAT1 prolongs the duration of IPSCs when synapses located close to each other are activated synchronously (Overstreet and Westbrook, 2003); the resulting prolongation of IPSCs increases the probability of IPSC summation and enhances the total efficacy of IPSC trains. The upregulation of the postsynaptic GABAA receptors that contain α2-subunits would be expected to increase the efficacy of the GABA that is released from chandelier neurons. Thus, the combined reduction of PV and GAT1 proteins in chandelier cell axon cartridges, and the upregulation of postsynaptic GABAA receptors, would act synergistically to increase the efficacy of GABA neurotransmission at pyramidal neuron axon initial segments during the types of repetitive neuronal activity associated with working memory. However, the persistence of cognitive impairments in individuals with schizophrenia suggests that these neuroplastic changes in GABA neurotransmission from chandelier neurons are insufficient as compensatory responses. Alternatively, it is possible that compensation at chandelier cell synapses is not effective because additional interneuron subclasses are also functionally deficient in schizophrenia (Lewis et al, 2005).
Are PV-containing Basket Cells also Altered in Schizophrenia?
The other major subclass of PV-containing GABA neurons are basket (or wide arbor) cells whose axons target the cell body and proximal dendritic spines and shafts of pyramidal neurons. Although more difficult to directly assess, similar presynaptic and postsynaptic alterations may also be present in the inputs of PV-containing, basket neurons to the perisomatic region of pyramidal neurons. For example, the density of PV-immunoreactive puncta, possibly the axon terminals of wide arbor neurons (Erickson and Lewis, 2002), is reduced in the middle layers, and not in the superficial layers, of the DLPFC of subjects with schizophrenia (Lewis et al, 2001a), paralleling the laminar pattern of decreased PV mRNA expression in schizophrenia (Hashimoto et al, 2003). Furthermore, the increased density of GABAA receptors in the DLPFC of subjects with schizophrenia found in ligand-binding studies (Benes et al, 1996; Hanada et al, 1987) was most prominent at pyramidal neuron cell bodies (Benes et al, 1996). Together, these data suggest that the perisomatic inhibitory input from both PV-containing chandelier and basket neurons is reduced in schizophrenia.
What are the Consequences of Altered GABA Neurotransmission in DLPFC PV-containing Neurons in Schizophrenia?
How could reduced GABA signaling from PV-containing GABA neurons to the perisomatic region of pyramidal neurons in the DLPFC contribute to the pathophysiology of working memory dysfunction? First, it is well established that the activity of DLPFC GABA neurons is essential for normal working memory function in monkeys (Rao et al, 2000; Sawaguchi et al, 1989). Second, PV-positive GABA neurons and pyramidal neurons share common sources (eg thalamic afferents) of excitatory input (Melchitzky et al, 1999). The resulting feedforward, disynaptic inhibition limits the time window for the summation of excitatory inputs required to evoke pyramidal neuron firing (Pouille and Scanziani, 2001). Third, both chandelier and basket neurons target multiple pyramidal neurons (Peters et al, 1982), enabling them to use this timing mechanism to synchronize the activity of local populations of pyramidal neurons (Klausberger et al, 2003). Finally, networks of PV-positive GABA neurons, formed by both chemical and electrical synapses, give rise to oscillatory activity in the γ band range, the synchronized firing of a neuronal population at 30–80 Hz (Whittington and Traub, 2003). Interestingly, γ band oscillations in the human DLPFC increase in proportion to working memory load (Howard et al, 2003), and in subjects with schizophrenia, prefrontal γ band oscillations are reduced bilaterally during a working memory task (Cho et al, 2006). Thus, a deficit in the synchronization of pyramidal cell firing, resulting from impaired regulation of pyramidal cell networks by PV-positive GABA neurons, may contribute to reduced levels of induced γ band oscillations, and consequently to impairments in cognitive tasks that involve working memory in subjects with schizophrenia (Lewis et al, 2005).
The decreased GAD67 mRNA expression in schizophrenia is not restricted to the DLPFC, with similar observations reported in studies of the anterior cingulate and temporal cortices (Akbarian and Huang, 2006). In addition, disturbances in γ oscillations evoked with auditory and visual stimuli have been reported in other cortical regions (Spencer et al, 2004) in subjects with schizophrenia. Thus, a conserved alteration in GABA neurotransmission across cortical regions could underlie a common abnormality in γ oscillations that is associated with different clinical features of schizophrenia. Consistent with this interpretation, a recent study found the same pattern of altered GABA-related transcripts across four different cortical regions in subjects with schizophrenia (Hashimoto et al, 2007b).
Is the Output of Chandelier Neurons Inhibitory or Excitatory?
Chandelier neurons have been considered to be powerful inhibitors of pyramidal cell output, exercising ‘veto power’ by virtue of the close proximity of their synaptic inputs to the site of action potential generation in pyramidal neurons. Like other cortical GABA neurons, the effect of GABA released from chandelier neuron axon terminals is mediated by binding to postsynaptic GABAA receptors, which results in the opening of chloride ion channels. In the adult brain, high expression of the potassium–chloride co-transporter (KCC2) results in the extrusion of chloride from the cell (Stein and Nicol, 2003). Thus, when GABAA receptors are activated, chloride ions flow into the cell along a concentration gradient, resulting in hyperpolarization of the membrane, and a reduced probability of cell firing. However, a recent study found that KCC2, while readily detectable in the cell body of adult pyramidal neurons, was apparently absent in the axon initial segment. Consistent with this observation, Szabadics et al (2006) found that the release of GABA from chandelier neuron axon terminals resulted in depolarization of pyramidal cells in an in vitro slice preparation. In fact, the chandelier cell-mediated depolarization was so powerful that in ∼50% of the cases in which a single chandelier cell was stimulated, the postsynaptic pyramidal cell was depolarized to the point of firing an action potential. In contrast, excitatory input from a single pyramidal neuron can rarely, if ever, cause a postsynaptic neuron to fire.
The findings of this study are quite striking, and suggest that the scenario described above for the physiological consequences of altered GABA neurotransmission in chandelier neurons in schizophrenia might need to be reconsidered. For example, if chandelier cell inputs to pyramidal neurons are usually depolarizing, then reduced synthesis of GABA in these neurons could result in a marked decrease in the excitatory output of pyramidal neurons that project to other cortical regions or subcortical sites. Alternatively, it is possible that instead of providing a substantial fraction of background excitatory synaptic input over time, chandelier neurons excite pyramidal cells only during specific patterns of network activity. For instance, by virtue of the divergent connections of a given chandelier cell to several hundred pyramidal cells (and assuming that every pyramidal cell receives chandelier cell inputs), then excitatory input from chandelier cells could provide a strong synchronization signal during network oscillations. However, the findings of the Szabadics et al's (2006) study, while awaiting replication, also need to be integrated with observations in the hippocampus of living animals showing that during θ oscillations the maximal activity of chandelier neurons is perfectly out of phase with that of pyramidal neurons (Klausberger et al, 2003), indicative of a powerful inhibitory timing effect of chandelier cells.
Are Other Populations of GABA Neurons Altered in Schizophrenia?
Although the findings reviewed above provide convergent evidence for alterations in PV-containing GABA neurons in schizophrenia, they also suggest that these neurons cannot account for all of the observed findings in postmortem studies. For example, the levels of GAD67 and GAT1 mRNAs are reduced to comparable degrees in layers 2–5 (Volk et al, 2000, 2001), even though the density of PV neurons is much greater in layers 3 and 4 than in layers 2 and 5 (Condé et al, 1994). In addition, PV mRNA expression was reduced in layers 3 and 4, but not in layers 2 and 5, in subjects with schizophrenia (Hashimoto et al, 2003). Together, these findings suggest that one or more populations of GABA neurons in layers 2 and 5, which express neither PV nor CR, is altered in schizophrenia.
A recent study using a customized cDNA microarray with enhanced sensitivity and specificity for 85 GABA-related transcripts found that, in addition to decreased expression of GAD67 and GAT1 mRNAs, the tissue concentrations of the mRNAs for three neuropeptides (SST, neuropeptide Y (NPY) and cholecystokinin (CCK)) were also reduced in the DLPFC of subjects with schizophrenia (Hashimoto et al, 2007a; Figure 3). These findings were confirmed by quantitative real-time PCR and/or in situ hybridization, and they are consistent with previous reports of decreased CCK peptide (Gabriel et al, 1996) and mRNA levels (Virgo et al, 1995) in other frontal areas. In the cortex, SST is expressed by GABA neurons that do not express PV or CR (González-Albo et al, 2001), and a subset of SST-containing neurons largely overlaps with the majority of NPY-containing neurons (Hendry et al, 1984; Kubota et al, 1994). Because SST- and NPY-containing neurons are predominantly located in layers 2 and 5 (Hendry et al, 1984; Kubota et al, 1994), alterations in these neurons could contribute to the deficits in GAD67 and GAT1 mRNA expression in these layers (Volk et al, 2000). Consistent with this interpretation, the gene expression differences between subjects with schizophrenia and control subjects were highly positively correlated for GAD67, SST, and NPY, suggesting that GAD67 mRNA expression is also decreased in the subset of GABA neurons that express both SST and NPY. Because SST- and NPY-containing neurons selectively target distal dendrites of pyramidal neurons (DeLima and Morrison, 1989; Hendry et al, 1984; Kawaguchi and Kubota, 1996), these coordinated gene expression changes suggest that GABA neurotransmission is altered at the dendritic domain of pyramidal neurons in the DLPFC of subjects with schizophrenia.

Schematic summary of alterations in GABA-mediated circuitry in the dorsolateral prefrontal cortex (DLPFC) of subjects with schizophrenia. Altered GABA neurotransmission by parvalbumin (PV)-containing neurons (green) is indicated by gene expression deficits in these neurons and associated changes in their synapses, a decrease in GAT1 expression in their terminals and an upregulation of GABAA receptor α2-subunit at the axon initial segments of pyramidal neurons (lower enlarged square). Decreased gene expression for both somatostatin (SST) and neuropeptide Y (NPY) indicates alterations in SST and/or NPY-containing neurons (blue) that target the distal dendrites of pyramidal neurons. Decreased cholecystokinin (CCK) mRNA levels indicate an alteration of CCK-containing large basket neurons (purple) that represent a separate source of perisomatic inhibition from PV-containing neurons. Gene expression in CR-containing GABA neurons (red) does not seem to be altered. Other neurons, such as PV-containing basket neurons, are not shown because the nature of their involvement in schizophrenia is unclear. G, generic GABA neuron; P, pyramidal neuron. Adapted from Hashimoto et al (2007a).
CCK is heavily expressed in GABA neurons that do not contain either PV or SST (Kawaguchi and Kondo, 2002; Lund and Lewis, 1993). The expression changes of CCK and GAD67 in the DLPFC of subjects with schizophrenia were highly correlated, suggesting a deficit of GABA synthesis in CCK-containing GABA neurons. Interestingly, the axon terminals of CCK-containing large basket neurons, which target selectively pyramidal neuron cell bodies, contain type I cannabinoid receptors (CB1R) (Eggan and Lewis, 2007). The activation of these receptors by exogenous cannabinoids suppresses GABA release (Bodor et al, 2005; Katona et al, 1999). Thus, exposure to cannabis could exacerbate an intrinsic deficit in GABA synthesis in these neurons, providing a mechanism by which cannabis use increases the risk for, and the severity of, schizophrenia (Fergusson et al, 2006). In addition, preliminary evidence of reduced mRNA and protein levels of CB1R in the DLPFC of subjects with schizophrenia suggests that downregulation of this receptor may represent a compensatory response to reduce the ability of endogenous cannabinoids to decrease GABA release from CCK/CB1R-containing axon terminals (Eggan et al, 2007). Interestingly, CCK/CB1R- and PV-containing basket cells provide convergent sources of perisomatic inhibition to pyramidal neurons that play specific roles in shaping network activity, including complementary roles in regulating γ band oscillations (Hajos et al, 2000). Thus, alterations in CCK-containing basket cells could also contribute to impaired γ oscillations in schizophrenia.
Could the Alterations in the Affected PV-containing GABA Neurons Reflect Disturbances in the Postnatal Maturation of their Synapses?
Of the subpopulations of GABA neurons that are affected in schizophrenia, the development of PV-containing neurons has been best studied in the primate DLPFC. For example, the density of the PV-positive axon terminals of basket neurons changes markedly during postnatal development (Erickson and Lewis, 2002), increasing steadily in an almost linear fashion from 1 month of age to adulthood (Figure 4). These developmental changes in the number of PV-positive boutons most likely reflect a shift in the detectability of basket axon terminals with immunocytochemical techniques, secondary to a change in the concentration of PV protein within the terminals, since the total number of inhibitory synapses, and the axonal arbors of basket cells specifically, appear to remain relatively constant over this same period of development (Bourgeois et al, 1994; Lund and Lewis, 1993).

Postnatal development of chandelier neuron inputs to pyramidal neurons in monkey dorsolateral prefrontal cortex (DLPFC). The axon terminals of chandelier neurons are vertical arrays of boutons (cartridges) that are immunoreactive for parvalbumin (PV) or the GABA membrane transporter (GAT1) and synapse on the axon initial segment of pyramidal neurons (a). As shown in (b), the density of cartridges immunoreactive for either PV or GAT1 is low in the DLPFC of newborn monkeys, increases to reach a peak before the onset of puberty, and then declines markedly during adolescence (shaded area between 15 and 42 months of age) to adult levels (Cruz et al, 2003). These density changes in PV- and GAT1-immunoreactive cartridges seem to reflect developmental shifts in the concentration of these proteins (and thus in the detectability of cartridges) because cartridges are readily visualized with the Golgi technique over this same time period (Lund and Lewis, 1993). Postsynaptically, the detectability of the α2-subunit of the GABAA receptor in pyramidal neuron axon initial segment is high at birth, and then markedly declines during adolescence before stable adult levels are achieved. In contrast, the density of PV-immunoreactive varicosities, putative axon terminals from the basket class of PV-expressing interneurons that innervate the cell body and proximal dendrites of pyramidal neurons, rises during adolescence (Erickson and Lewis, 2002). Together, these developmental changes in markers of inhibition at the perisomatic region of pyramidal neurons suggest that the capacity to synchronize pyramidal neuron output in the primate DLPFC is in substantial flux until adulthood. Consequently, the protracted developmental time course of improvements in performance on working memory tasks (Crone et al, 2006; Diamond, 2002; Luna et al, 2004) might depend on both refinements in the number of excitatory connections among pyramidal neurons and changes in their proximal inhibitory inputs. These developmental changes during adolescence might contribute to unmasking the consequences of inherited abnormalities in the regulation of GABAergic neurotransmission and might help explain why certain life experiences during adolescence (eg stress or cannabis exposure) seem to increase the risk for schizophrenia (Lewis and Levitt, 2002). Adapted from Lewis et al (2005).
The chandelier class of PV-containing GABA neurons also exhibits developmental changes in the expression of biochemical markers. During postnatal development, the density of chandelier neuron axon cartridges immunoreactive for either PV or GAT1 changes markedly in monkey DLPFC (Cruz et al, 2003). Although the precise time course differs for the two markers, the density of labeled cartridges is low in the newborn, increases to reach a peak prior to the onset of puberty, and then declines markedly to adult levels (Figure 4). Because cartridges are readily visualized with the Golgi technique over this same time period (Lund and Lewis, 1993), the changes in PV- and GAT-immunoreactive cartridges likely reflect developmental shifts in the concentration of these proteins. Interestingly, the peak and subsequent decline in the density of labeled cartridges occur prior to the age when the peak density of labeled basket terminals is achieved, but appears concomitant with the pruning of intracortical excitatory connections (Woo et al, 1997b). Although the basis for this temporal coincidence remains to be investigated, it may be that the synchronous firing of a group of pyramidal cells, facilitated by inputs from chandelier neurons, is essential for promoting the maintenance of a subset of excitatory connections.
Postsynaptically, the influence of released GABA on the pyramidal cells targeted by chandelier neurons is determined by the particular type of GABAA receptor to which it binds. Interestingly, GABAA receptors undergo changes in subunit composition during cortical development. For example, the α2-subunit is most prevalent prenatally and is replaced in the majority of, but not all, GABAA receptors postnatally by the α1-subunit (Fritschy et al, 1994). In the adult cortex, the majority of GABAA receptors containing the α2-subunit are found in pyramidal cell axon initial segments (Loup et al, 1998). The detectability of GABAA α2-subunits at the axon initial segment undergoes substantial changes during postnatal development (Cruz et al, 2003). As shown in Figure 4, the density of pyramidal cell axon initial segments immunoreactive for the α2-subunit is very high in the postnatal period, and then steadily declines through adolescence into adulthood. Because GABA receptors including the α2-subunit have a higher affinity for GABA, faster activation times, and slower deactivation times than receptors containing the α1-subunit (Lavoie et al, 1997), this decrease in the density of α2-labeled axon initial segments may be interpreted as a change in the speed and efficacy of GABAergic transmission at the axon initial segment during postnatal maturation, rather than a reduction in the number of GABAergic synapses onto the axon initial segment, because the total number of inhibitory synapses remains constant over this same time period (Bourgeois et al, 1994).
This complex and protracted postnatal maturation of the inputs from PV-containing GABA neurons in the primate DLPFC provides a number of opportunities for any disturbances, even subtle ones, to have their effects amplified as they alter the trajectories of the developmental events that follow. In particular, the marked developmental changes in the axon terminals of PV-containing basket and chandelier neurons, and their postsynaptic receptors, during the perinatal period and adolescence raises the possibility that the alterations in schizophrenia of these markers reflect a disturbance in these patterns of development. These temporal correlations may explain how a range of environmental factors (eg labor-delivery complications, urban place of rearing, and marijuana use during adolescence) are all associated with increased risk for the appearance of schizophrenia later in life. Although it seems unlikely that the GABA-related disturbances in schizophrenia represent an arrest of development, they may reflect an alteration of DLPFC circuitry that makes it unable to support higher levels of working memory load, rendering the impaired performance in schizophrenia analogous to the immature levels of working memory function seen in children (Crone et al, 2006; Diamond, 2002; Luna et al, 2004).
NEUROPLASTICITY OF MODULATORY CONNECTIONS IN SCHIZOPHRENIA
Evidence of Altered Dopamine Neurotransmission in the DLPFC in Schizophrenia
In addition to disturbances in excitatory and inhibitory connectivity in the DLPFC, alterations in modulatory systems, such as dopamine (DA), have also been found in schizophrenia. The positive or psychotic features of the illness were originally related to a functional excess of DA neurotransmission because (1) DA agonists, such as amphetamine, can induce a schizophrenia-like psychosis, and (2) antipsychotic medications have antagonistic activity at DA receptors of the D2 subtype (Carlsson, 2006). Indeed, the efficacy of classical antipsychotics against the positive symptoms of schizophrenia is strongly correlated with their potency at D2 receptors in subcortical structures (Carlsson, 2006). In contrast, impairments of cognitive function in schizophrenia might be related to a deficit in DA signaling in the DLPFC (Davis et al, 1991; Weinberger, 1987). For example, working memory performance is markedly impaired by depletion of DA (Brozoski et al, 1979), and by blockade of D1 receptor-mediated DA signaling (Sawaguchi and Goldman-Rakic, 1991, 1994), in the monkey DLPFC. Potential mechanisms underlying both an increase in DA input to subcortical sites and a simultaneous decrease in the DA signal in the DLPFC in schizophrenia have been reviewed elsewhere (Carlsson, 2006; Lewis and Gonzalez-Burgos, 2006). Briefly, it is thought that prefrontal pyramidal neurons may directly excite mesocortical DA neurons in the ventral mesencephalon and indirectly inhibit mesostriatal DA cells through activation of GABAergic neurons in mesencephalic cell nuclei (Lewis and Gonzalez-Burgos, 2006). Such opposing effects of the prefrontal projections on DA cells are in line with the idea that reduction in PFC cell activity leads to an excess of DA receptor activation at subcortical nuclei that is prevented by antipsychotics that antagonize D2 receptors. However, decreased DA input to cortex by reduced PFC cell activity could be at least partially counteracted by D1 receptor agonist administration.
In the primate DLPFC, D1 receptors are the most abundant DA receptor subtype (Goldman-Rakic et al, 1990) and mediate most of the cellular effects of DA in this region (González-Burgos et al, 2002; Gonzalez-Burgos et al, 2005; Henze et al, 2000; Kroner et al, 2007). Moreover, D1 receptors regulate sustained firing of DLPFC neurons during the delay phase of delayed-response tasks that require working memory (Sawaguchi, 2001; Williams and Goldman-Rakic, 1995). Sustained firing of DLPFC neurons is thought to be the cellular basis of information storage in the short-term buffers associated with working memory (Goldman-Rakic, 1995). DA input to the DLPFC may be necessary, in addition, for proper DLPFC neuron activity during other stages of the working memory process. For instance, certain effects of DA on neurons from the rat medial PFC suggest an important role of DA in gating the information loaded into working memory buffers (Rotaru et al, 2007; Seamans and Yang, 2004); Yang et al, 1999).
Postmortem studies indicate that the total length of tyrosine hydroxylase (TH)- and DA transporter-containing axons is significantly reduced in the DLPFC of subjects with schizophrenia (Akil et al, 1999). These findings suggest that the cortical DA signal might be diminished in schizophrenia, due to a reduced content of TH per axon, to a reduced density of DA axons or to both. However, schizophrenia does not seem to be associated with a substantial loss of midbrain DA neurons (Bogerts et al, 1983), although the DA cells in subjects with schizophrenia have reduced soma volume (Bogerts et al, 1983). Because reductions in soma volume are usually correlated with restricted axonal projections (Gilbert and Kelly, 1975; Lund et al, 1975), decreased DA signaling in the DLPFC in schizophrenia might thus result, at least in part, from a reduced DA innervation of these cortical areas. Consistent with the idea of altered midbrain DA neuron function in schizophrenia, a recent study reported reduced levels of tyrosine hydroxylase protein in midbrain DA cells in schizophrenia (Perez-Costas et al, 2007).
What Factors might Account for a Deficit in DA Signaling in the DLPFC?
The morphological alterations of DA neurons in schizophrenia, including reduced innervation of the DLPFC, might be the consequence of sustained hypoactivity of these neurons. DA cell activity is under control of multiple synaptic input pathways including a strong inhibitory input originating in subcortical structures (Grace et al, 2007; White, 1996). Glutamatergic inputs from subcortical structures, from the prefrontal cortex and, indirectly, from the hippocampus, provide excitation to DA cells (Grace et al, 2007; White, 1996). In rodents, excitatory projections from the PFC are thought to be an essential source of glutamate-mediated excitation to the PFC-projecting DA neurons (Abi-Dargham and Moore, 2003; Sesack and Carr, 2002). Sustained hypoactivity of PFC pyramidal neurons in schizophrenia may result from reduced NMDA-mediated signaling or from other changes in the excitatory inputs to these cells (see above). If such alterations ultimately affect the output of cortical pyramidal cells projecting to midbrain DA cell nuclei, then DLPFC cell hypoactivity may lead to persistently decreased activation of DA cells. However, a recent study suggested that in primates, unlike in rodents, prefrontal projections to midbrain DA cells are sparse (Frankle et al, 2006).
Altered DA input to the DLPFC in schizophrenia may also result from disturbances in the development of DA innervation. In the monkey DLPFC, the density of TH-containing DA fibers (Rosenberg and Lewis, 1995) and the number of TH-positive appositions onto pyramidal cell dendrites (Lambe et al, 2000) increase gradually during postnatal development, reaching a maximum during adolescence. In contrast, tissue levels of DA and TH protein are high in the primate DLPFC shortly after birth, but decrease markedly during the early postnatal period, prior to adolescence (Goldman-Rakic and Brown, 1982; Weickert et al, 2007). Thus, between childhood and adolescence, the DA fibers innervating the DLPFC are in a highly dynamic state, adjusting the levels of TH expression and increasing the density of axon branches and varicosities in target cortical areas. The mechanisms regulating the innervation of cortical areas by DA axons during development are not well understood, but neurotrophic factors such as the glia-derived neurotrophic factor (GDNF) and BDNF seemed to be involved. For instance, GDNF expression by cortical tissue promotes its innervation by DA axons, regulating the density of DA fibers in the adult state (Kholodilov et al, 2004). In addition, experiments with BDNF knockout mice suggest that BDNF stimulates TH expression by midbrain DA neurons (Baquet et al, 2005). Alterations of BDNF-mediated signaling in schizophrenia are well documented (Hashimoto et al, 2005; Weickert et al, 2005), but no similar changes have been reported for GDNF.
In contrast to subcortical structures, the DA transporter in cortical axons is located distant from release sites, limiting the ability of these transporters to regulate extracellular DA levels (Lewis et al, 2001b). In contrast, inhibition of the DA-degrading enzyme catechol-O-methyl transferase (COMT) significantly increases prefrontal DA levels (Tunbridge et al, 2004b), suggesting that alterations in COMT levels could contribute to altered DA signaling in schizophrenia. The levels of COMT mRNA and protein do not appear to be altered in schizophrenia (Matsumoto et al, 2003; Tunbridge et al, 2004a), although one study found significantly increased levels of a membrane-bound form of COMT in both schizophrenia and bipolar disorder (Tunbridge et al, 2006). The COMT gene is located in a chromosomal region associated with increased risk for schizophrenia, and its allelic variants code for enzymes with marked differences in catalytic activity (Chen et al, 2004). In particular, the Val158Met COMT allele was hypothesized to confer susceptibility for schizophrenia, because its presence is associated with lower DA levels and less robust cognitive performance in normal subjects (Egan et al, 2001). Consistent with this prediction, the presence of the Val allele, which codes for an isoform of COMT protein with higher enzymatic activity, is associated with higher levels of TH mRNA in mesencephalic DA neurons in healthy human subjects (Akil et al, 2003). However, studies of the association between COMT gene variants and schizophrenia have been inconclusive (Barnett et al, 2007; Craddock et al, 2006; Meyer-Lindenberg et al, 2006; Williams et al, 2007). Interestingly, COMT is mostly an intracellular cytosolic soluble protein, and although membrane-bound forms can be isolated (Tunbridge et al, 2006), these are probably associated with the intracellular side of the plasma membrane or with internal cell membranes (Mannisto and Kaakkola, 1999). Therefore, extracellular DA must be transported through the plasma membrane before becoming a substrate of COMT enzymatic activity, raising the question of potential interactions between genetic variants of COMT and the DA transporter. Indeed, allelic variants of the DA transporter gene are associated with different levels of transporter protein expression and may have significant effects on cortical physiology (Demiralp et al, 2007). Thus, certain genotypic combinations of COMT and DA transporter alleles might be associated with greater DA metabolism, decreased DA levels, deficits in cognitive functions and possibly an increased risk of developing schizophrenia. Although in some studies, the effects of both COMT and DA transporter gene polymorphisms on cognitive function have been assessed (Rybakowski et al, 2006), to our knowledge no studies have reported significant interactions between COMT and DA transporter gene variants. One possibility is that, because COMT expression levels in DA neurons are relatively low (Kastner et al, 1994; Lundstrom et al, 1995), the availability of DA to be inactivated by COMT is dependent on DA transport through the membrane of cortical cells that express higher levels of COMT (eg postsynaptic neurons or glia), and that may express non-specific membrane transporters with the ability to uptake DA. It is also important to note that COMT metabolizes norepinephrine which heavily innervates the primate DLPFC (Lewis and Morrison, 1989) and influences working memory function (Arnsten and Goldman-Rakic, 1985).
What are the Functional Consequences of Decreased DA Signaling in the DLPFC?
A combination of DA cell hypoactivity, decreased cortical DA innervation, and increased DA turnover in the DLPFC would lead to markedly reduced extracellular DA levels in the DLPFC in schizophrenia, which in turn may produce DA receptor hypostimulation. A potential consequence of long-term deficits in DA receptor stimulation, as indicated by some animal studies (Butkerait and Friedman, 1993; Davidoff et al, 2000; Guo et al, 2003; Tassin et al, 1986), is a compensatory increase in the DA receptor levels. Because antipsychotics decrease the levels of DA receptors in monkey and human DLPFC (Hirvonen et al, 2006; Lidow and Goldman-Rakic, 1994, 1997), assessment of effect of disease on DA receptor binding in the DLPFC of schizophrenic subjects should be performed, ideally, in drug naive patients. A positron emission tomography study revealed an increase in the binding of NNC112, a DA D1 receptor ligand, in drug-free and drug-naive patients, consistent with a compensatory D1 receptor upregulation in response to reduced DA levels (Abi-Dargham et al, 2002). Using SCH23390 instead of NNC112 to label D1 receptors in vivo, a separate study found decreased levels of D1 receptors in the DLPFC of drug naive and drug-free schizophrenia patients (Okubo et al, 1997). A third study found no differences between the in vivo binding of SCH23390 to the DLPFC of schizophrenics and control subjects (Karlsson et al, 2002). Further research is necessary to determine the factors underlying these discrepant findings. Interestingly, sustained DA depletion differentially affects NNC112 and SCH23390 binding in the rat brain, elevating the in vivo binding of the first but not the second ligand (Guo et al, 2003).
The degree of D1 receptor upregulation in schizophrenia was found to be inversely related to working memory performance (Abi-Dargham et al, 2002). However, D1 upregulation does not contribute to working memory dysfunction since D1 receptor antagonists worsen cognitive deficits in schizophrenia (Abi-Dargham and Moore, 2003). These results are consistent with the idea that D1 upregulation is a compensatory, but insufficient, response to the DA deficit in the DLPFC. In other studies, however, the decrease in cognitive function was associated with a lower D1 receptor binding (Okubo et al, 1997) or the magnitude of negative symptoms was not correlated to D1 receptor levels (Karlsson et al, 2002). Further research is necessary to determine the relationship between D1 receptor levels and cognitive/negative symptoms in schizophrenia, to establish the possibility of targeting D1 for treatment with novel drugs as well as to determine potential side effects of classic antipsychotics on cognitive function.
The cellular mechanisms of DA modulation of DLPFC neuron activity are important to understand the functional consequences of decreased DA signaling in the DLPFC in schizophrenia as well as the compensatory responses triggered by the DA deficit. DA modulatory connections differ substantially from glutamate and GABA synaptic connections because DA acts exclusively via G-protein-coupled receptors (Missale et al, 1998; Neve et al, 2004) unlike glutamate and GABA, which also act via fast ligand-gated channels. Thus, DA does not act via fast, point-to-point synaptic transmission, but in a slow and diffuse manner consistent with volume transmission mechanisms (Paspalas and Goldman-Rakic, 2004). Indeed, D1 receptors are found in dendritic shafts and spines (Bergson et al, 1995; Paspalas and Goldman-Rakic, 2004, 2005; Smiley et al, 1994), and in glutamate axonal boutons (Paspalas and Goldman-Rakic, 2005), most of which are not associated with DA terminals. DA signaling via volume transmission suggests that changes in DA degradation by COMT or in the uptake of DA by DA transporters could lead to significant changes in the amount of DA available to activate D1 receptors. As noted above, robust changes in the levels of COMT, consistent with either deficiency or compensation, have not been found in schizophrenia. In contrast, the decrease in detectable DA transporter-containing fibers in the DLPFC in schizophrenia (Akil et al, 1999), may reflect to some extent a compensatory decrease in DA uptake that favors volume transmission.
Physiological studies have focused on the DA modulation of intrinsic membrane properties and cellular excitability and of glutamate- and GABA-mediated transmission. Application of DA or D1 agonists to monkey DLPFC slices does not affect the membrane potential of pyramidal neurons (González-Burgos et al, 2002; Henze et al, 2000). In contrast, DA consistently depolarizes interneurons of the fast-spiking subclass, but not interneurons of other subtypes (Kroner et al, 2007). Because most fast-spiking cells contain PV, and most PV cells are fast-spiking, selective regulation of fast-spiking interneuron activity by DA is consistent with studies showing that PV neurons, but not calretinin-positive cells, receive synaptic inputs from DA axons (Sesack et al, 1998) and preferentially express D1 receptors (Muly et al, 1998). Probably as a consequence of increased FS cell excitability, D1 receptor activation also increases the frequency of action potential-dependent inhibitory synaptic currents in pyramidal cells (Kroner et al, 2007). Because the expression of GAD67 mRNA in interneurons is activity-dependent (Benson et al, 1994) and because DA upregulates the firing of fast-spiking cells (Kroner et al, 2007), a deficient DA signal in DLPFC may contribute to a decrease in fast-spiking cell activity and thus decreased GAD67 expression in these neurons (see Figure 5).

Summary of the putative causes and consequences of altered dopamine (DA) signaling in the dorsolateral prefrontal cortex (DLPFC) in schizophrenia. A decrease in glutamatergic excitatory input onto mesencephalic DA cells may result from hypoactivity in pyramidal cells (P) located in the DLPFC or in other cortical areas that are a source of excitatory input to DA cells. Reduced excitatory input may produce sustained hypoactivity of DA cells providing DA input to the DLPFC, which may result in both morphological and biochemical changes in the DA cells. For instance, in schizophrenia the size of DA neuron cell bodies has been reported to be reduced and might be associated with a lower number of DA fibers innervating the DLPFC. In addition, the amount of tyrosine hydroxylase (TH) in the mesencephalic DA cell nuclei has been reported to be reduced in schizophrenia as is the detectability of TH and of dopamine transporter (DAT)-immunoreactive DA axons in the DLPFC. Decreased DA innervation and reduced DA levels in the DLPFC may lead to compensatory, but functionally insufficient, upregulation of D1 receptors in pyramidal cells or GABA neurons or both. Because D1 receptor activation increases the activity of parvalbumin (PV)-positive, fast-spiking GABA neurons, reduced D1-mediated signaling may reduce the activity of these neurons and lead to activity-dependent downregulation of the expression of GAD67 and PV.
Although the interneuron depolarization and the increase in inhibitory currents produced by DA suggests net inhibitory effects in the primate DLPFC network, DA also changes the response of pyramidal neurons to excitatory currents, although in complex ways. In vitro, DA increases the excitability of primate DLPFC pyramidal cells with a delayed time course (González-Burgos et al, 2002; Henze et al, 2000) that is consistent with other delayed D1 receptor-mediated effects (Seamans and Yang, 2004). However, more detailed studies of the effects of DA receptor activation on rat medial PFC pyramidal neurons revealed effects consistent with an early decrease in excitability, followed by a delayed enhancement (Seamans and Yang, 2004). The mechanisms underlying short-term and long-lasting effects of DA receptor activation, as well as their relative importance in vivo, remain to be determined. In addition to such temporally biphasic effects in vitro, DA seems to have effects in vivo that are biphasic as a function of its extracellular concentration or occupancy of DA receptors. More specifically, DA elicits effects on monkey DLPFC cell firing that follow an ‘inverted-U’ dose–response relationship (Vijayraghavan et al, 2007; Williams and Goldman-Rakic, 1995). However, such ‘inverted-U’ effects in vivo seem to result from complex modulatory effects and not simply from modulation of monkey DLPFC pyramidal cell excitability such as that found in vitro (González-Burgos et al, 2002; Henze et al, 2000). For instance, low levels of DA receptor stimulation do not generally enhance the firing of DLPFC cells in vivo, but selectively increase the firing of neurons that are tuned to preferred stimuli during tasks that require working memory (Vijayraghavan et al, 2007; Williams and Goldman-Rakic, 1995). Therefore, the effects of DA receptor activation on the output of DLPFC pyramidal cells most likely result from complex network effects.
Enhancement of tuned firing of DLPFC neurons by low levels of DA receptor stimulation during cognitive tasks is consistent with the idea that cognitive deficits in schizophrenia result, at least in part, from decreased DA input to the DLPFC. Moreover, D1 receptor upregulation in schizophrenia might partially compensate for such deficits in working memory-related DLPFC activity. Because high levels of DA receptor stimulation, via this ‘inverted-U’ model, disrupt tuned firing of DLPFC neurons, D1 receptor-based treatment of cognitive deficits in schizophrenia should consider the changes in D1 receptor levels produced by compensatory brain mechanisms and also the changes in D1 receptors induced by antipsychotic medications. For instance, PET-based assessments of the degree of D1 receptor upregulation in individual patients may help in guiding therapy to maximize the possibility that therapeutic compounds are combined with the compensatory plasticity in D1 receptor levels in a manner that restores optimal levels of D1 receptor stimulation.
FUTURE RESEARCH DIRECTIONS
The findings reviewed above indicate that the dysfunction of the DLPFC and associated working memory impairments in schizophrenia are likely the result of a complex set of alterations in glutamate, GABA, and DA neurotransmission. The existing data suggest that some of these alterations are deleterious causes or consequences of disturbances in the functional architecture of the DLPFC and interconnected brain regions, whereas others are best explained as compensatory responses. In each case, they reflect the neuroplasticity of DLPFC circuitry in a disease state.
How do these changes arise in the context of neuroplastic responses? Clearly, the nature of the relationships of these alterations within and across excitatory, inhibitory, and modulatory circuits requires further investigation. For example, it remains unclear whether these changes define diversity and/or variance in the disease process(es) that underlie the clinical syndrome we call schizophrenia. That is, do some individuals have predominately disturbances in one of these systems, but not in another, yet with the frequency of alterations in each system common enough to be detected in cohorts of subjects identified by the presence of the clinical syndrome and not specific phenotypic features? It is also critical to refine the cellular and circuitry specificity of the molecular alterations in the illness. For example, as summarized in the section on Excitatory Cortical Connections, morphological evidence of alterations in excitatory circuits in the DLPFC appear to be most pronounced in deep layer 3 pyramidal neurons. However, further explication of these findings requires the availability of a panel of molecular markers that define distinct subsets of pyramidal neurons based on their functional and connectional properties. In the case of the disturbances in GABA neurotransmission, a fuller understanding of whether changes in other GABAA receptor subunits (Hashimoto et al, 2007a) or GABAB receptor subunits (Zhao et al, 2007) consistently occur in the illness, and if so, whether they occur in a cell type and circuit-specific manner may inform their potential utility as therapeutic targets.
Understanding these issues is necessary for a pathophysiologically based rationale for the development of novel and selective pharmacotherapic interventions for the illness, that is, any molecule identified as a drug target must be understood in the cellular and circuitry context of the DLPFC, as well as other brain regions that express the molecule of interest. In addition, understanding whether the disease-related change in the targeted molecule is a consequence or compensation in the disease process has important implications both for the nature of activity of the drugs designed against that target, and perhaps for the potential therapeutic value of the target. For example, is the neuroplastic capacity of cortical circuitry sufficiently limited that pharmacological augmentation of a compensatory response is feasible? If so, is such an approach more likely to be effective than attempts to reverse detrimental consequences when the pathological cause is still present? Furthermore, given the marked developmental changes that occur in each of these systems during adolescence, this type of pharmacological intervention may have particular value as an intervention strategy for high-risk adolescents in the prodromal phase of the illness. However, the effectiveness and safety of such interventions requires a complete understanding of the maturation of these neural circuits, of the functional consequences of these circuitry changes and of the vulnerability of these developmental processes to pharmacological agents.
References
Abi-Dargham A, Mawlawi O, Lombardo I, Gill R, Martinez D, Huang Y et al (2002). Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci 22: 3708–3719. This important study provided an in vivo evidence of altered dopamine neurotransmission in the prefrontal cortex in schizophrenia.
Abi-Dargham A, Moore H (2003). Prefrontal DA transmission at D1 receptors and the pathology of schizophrenia. Neuroscientist 9: 404–416.
Adler CM, Malhotra AK, Elman I, Goldberg T, Egan M, Pickar D et al (1999). Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am J Psychiatry 156: 1646–1649.
Akbarian S, Huang HS (2006). Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev 52: 293–304.
Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney Jr WE et al (1995). Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry 52: 258–266. This study provided the seminal evidence for a deficit of GABA synthesis in schizophrenia and that this alteration might be cell type-specific.
Akbarian S, Sucher NJ, Bradley D, Tafazzoli A, Trinh D, Hetrick WP et al (1996). Selective alterations in gene expression of NMDA receptor subunits in prefrontal cortex of schizophrenics. J Neurosci 16: 19–30.
Akil M, Kolachana BS, Rothmond DA, Hyde TM, Weinberger DR, Kleinman JE (2003). Catechol-O-methyltransferase genotype and dopamine regulation in the human brain. J Neurosci 23: 2008–2013.
Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR et al (1999). Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry 156: 1580–1589.
Alvarez VA, Sabatini BL (2007). Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci 30: 79–97.
Anderson SA, Classey JD, Condé F, Lund JS, Lewis DA (1995). Synchronous development of pyramidal neuron dendritic spines and parvalbumin-immunoreactive chandelier neuron axon terminals in layer III of monkey prefrontal cortex. Neuroscience 67: 7–22.
Arellano JI, Espinosa A, Fairen A, Yuste R, DeFelipe J (2007). Non-synaptic dendritic spines in neocortex. Neuroscience 145: 464–469.
Arion D, Unger T, Lewis DA, Mirnics K (2007). Molecular markers distinguishing supragranular and infragranular layers in the human prefrontal cortex. Eur J Neurosci 25: 1843–1854.
Arnsten AFT, Goldman-Rakic PS (1985). Alpha(2)-adrenergic mechanisms in prefrontal cortex associated with cognitive decline in aged nonhuman primates. Science 230: 1273–1276.
Baddeley A (1992). Working memory. Science 255: 556–559.
Baquet ZC, Bickford PC, Jones KR (2005). Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci 25: 6251–6259.
Barbas H (1992). Architecture and cortical connections of the prefrontal cortex in the Rhesus monkey. Adv Neurol 57: 91–115.
Barch DM (2006). What can research on schizophrenia tell us about the cognitive neuroscience of working memory? Neuroscience 139: 73–84.
Barch DM, Sheline YI, Csernansky JG, Snyder AZ (2003). Working memory and prefrontal cortex dysfunction: specificity to schizophrenia compared with major depression. Biol Psychiatry 53: 376–384.
Barnett JH, Jones PB, Robbins TW, Muller U (2007). Effects of the catechol-O-methyltransferase Val(158)Met polymorphism on executive function: a meta-analysis of the Wisconsin Card Sort Test in schizophrenia and healthy controls. Mol Psychiatry 12: 502–509.
Benes FM, Vincent SL, Marie A, Khan Y (1996). Up-regulation of GABA-A receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience 75: 1021–1031.
Benson DL, Huntsman MM, Jones EG (1994). Activity-dependent changes in GAD and preprotachykinin mRNAs in visual cortex of adult monkeys. Cereb Cortex 4: 40–51.
Bergson C, Mrzljak L, Smiley JF, Pappy M, Levenson R, Goldman-Rakic PS (1995). Regional, cellular, and subcellular variations in the distribution of D1 and D5 dopamine receptors in primate brain. J Neurosci 15: 7821–7836.
Bird ED, Spokes EGS, Iversen LL (1979). Increased dopamine concentration in limbic areas of brain from patients dying with schizophrenia. Brain 102: 347–360.
Black JE, Kodish IM, Grossman AW, Klintsova AY, Orlovskaya D, Vostrikov V et al (2004). Pathology of layer V pyramidal neurons in the prefrontal cortex of patients with schizophrenia. Am J Psychiatry 161: 742–744.
Blanpied TA, Ehlers MD (2004). Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol Psychiatry 55: 1121–1127.
Bodor AL, Katona I, Nyiri G, Mackie K, Ledent C, Hajos N et al (2005). Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J Neurosci 25: 6845–6856.
Bogerts B, Hantsch J, Herzer M (1983). A morphometric study of the dopamine-containing cell groups in the mesencephalon of normals, Parkinson patients, and schizophrenics. Biol Psychiatry 18: 951–969.
Bourgeois J-P, Goldman-Rakic PS, Rakic P (1994). Synaptogenesis in the prefrontal cortex of rhesus monkeys. Cereb Cortex 4: 78–96.
Broadbelt K, Byne W, Jones LB (2002). Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr Res 58: 75–81.
Brozoski TJ, Brown RM, Rosvold HE, Goldman PS (1979). Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkeys. Science 205: 929–932. This report provided the first evidence that the dopamine innervation of the primate prefrontal cortex is necessary for correct performance during working memory tasks.
Butkerait P, Friedman E (1993). Repeated reserpine increases striatal dopamine receptor and guanine nucleotide binding protein RNA. J Neurochem 60: 566–571.
Byne W, Buchsbaum MS, Mattiace LA, Hazlett EA, Kemether E, Elhakem SL et al (2002). Postmortem assessment of thalamic nuclear volumes in subjects with schizophrenia. Am J Psychiatry 159: 59–65.
Calabrese B, Wilson MS, Halpain S (2006). Development and regulation of dendritic spine synapses. Physiology (Bethesda) 21: 38–47.
Callicott JH, Mattay VS, Verchinski BA, Marenco S, Egan MF, Weinberger DR (2003). Complexity of prefrontal cortical dysfunction in schizophrenia: more than up or down. Am J Psychiatry 160: 2209–2215.
Cannon TD, Glahn DC, Kim J, Van Erp TG, Karlsgodt K, Cohen MS et al (2005). Dorsolateral prefrontal cortex activity during maintenance and manipulation of information in working memory in patients with schizophrenia. Arch Gen Psychiatry 62: 1071–1080.
Carlisle HJ, Kennedy MB (2005). Spine architecture and synaptic plasticity. Trends Neurosci 28: 182–187.
Carlsson A (2006). The neurochemical circuitry of schizophrenia. Pharmacopsychiatry 39 (Suppl 1): S10–S14.
Chen J, Lipska BK, Halim N, Ma QD, Matsumoto M, Melhem S et al (2004). Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet 75: 807–821.
Cho RY, Konecky RO, Carter CS (2006). Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc Natl Acad Sci USA 103: 19878–19883.
Collin T, Chat M, Lucas MG, Moreno H, Racay P, Schwaller B et al (2005). Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J Neurosci 25: 96–107.
Condé F, Lund JS, Jacobowitz DM, Baimbridge KG, Lewis DA (1994). Local circuit neurons immunoreactive for calretinin, calbindin D-28k, or parvalbumin in monkey prefrontal cortex: distribution and morphology. J Comp Neurol 341: 95–116.
Coyle JT (2004). The GABA-glutamate connection in schizophrenia: which is the proximate cause? Biochem Pharmacol 68: 1507–1514.
Craddock N, Owen MJ, O'Donovan MC (2006). The catechol-O-methyl transferase (COMT) gene as a candidate for psychiatric phenotypes: evidence and lessons. Mol Psychiatry 11: 446–458.
Crone EA, Wendelken C, Donohue S, van Leijenhorst L, Bunge SA (2006). Neurocognitive development of the ability to manipulate information in working memory. Proc Natl Acad Sci USA 103: 9315–9320.
Cruz DA, Eggan SM, Lewis DA (2003). Postnatal development of pre- and post-synaptic GABA markers at chandelier cell inputs to pyramidal neurons in monkey prefrontal cortex. J Comp Neurol 465: 385–400.
Cullen TJ, Walker MA, Parkinson N, Craven R, Crow TJ, Esiri MM et al (2003). A postmortem study of the mediodorsal nucleus of the thalamus in schizophrenia. Schizophr Res 60: 157–166.
Dailey ME, Smith SJ (1996). The dynamics of dendritic structure in developing hippocampal slices. J Neurosci 16: 2983–2994.
Davidoff SA, Chu HM, Benes FM (2000). Acute administration of SCH23390 increases D(1) receptors on nonpyramidal neurons in rat mPFC. Synapse 35: 173–181.
Davis KL, Kahn RS, Ko G, Davidson M (1991). Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry 148: 1474–1486.
DeFelipe J (1997). Types of neurons, synaptic connections and chemical characteristics of cells immunoreactive for calbindin-D28K, parvalbumin and calretinin in the neocortex. J Chem Neuroanat 14: 1–19.
DeLima AD, Morrison JH (1989). Ultrastructural analysis of somatostatin-immunoreactive neurons and synapses in the temporal and occipital cortex of the macaque monkey. J Comp Neurol 283: 212–227.
Demiralp T, Herrmann CS, Erdal ME, Ergenoglu T, Keskin YH, Ergen M et al (2007). DRD4 and DAT1 polymorphisms modulate human gamma band responses. Cereb Cortex 17: 1007–1019.
Diamond A (2002). Normal development of prefrontal cortex from birth to young adulthood: cognitive functions, anatomy and biochemistry. In: Stuss DT, Knight RT (eds). Principles of Frontal Lobe Function. Oxford University Press: London. pp 466–503.
Dorph-Petersen KA, Pierri JN, Wu Q, Sampson AR, Lewis DA (2007). Primary visual cortex volume and total neuron number are reduced in schizophrenia. J Comp Neurol 501: 290–301.
Dorph-Petersen K-A, Pierri JN, Sun Z, Sampson AR, Lewis DA (2004). Stereological analysis of the mediodorsal thalamic nucleus in schizophrenia: Volume, neuron number, and cell types. J Comp Neurol 472: 449–462.
Dracheva S, Marras SAE, Elhakem SL, Kramer FR, Davis KL, Haroutunian V (2001). N-methyl-D-aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia. Am J Psychiatry 158: 1400–1410.
Duan H, Wearne SL, Morrison JH, Hof PR (2002). Quantitative analysis of the dendritic morphology of corticocortical projection neurons in the macaque monkey association cortex. Neuroscience 114: 349–359.
Dudkin KN, Kruchinin VK, Chueva IV (2001). Neurophysiological correlates of delayed visual differentiation tasks in monkeys: the effects of the site of intracortical blockade of NMDA receptors. Neurosci Behav Physiol 31: 207–218.
Durstewitz D, Seamans JK (2006). Beyond bistability: biophysics and temporal dynamics of working memory. Neuroscience 139: 119–133.
Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE et al (2001). Effect of COMT Val 108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci USA 98: 6917–6922. This elegant study describes the potential role of COMT in the pathophysiology of frontal lobe dysfunction in schizophrenia and provides evidence supporting the hypothesis that deficient cortical dopamine neurotransmission could contribute to the cognitive disturbances of the illness.
Eggan SM, Hashimoto T, Lewis DA (2007). Alterations in CB1 receptor MRNA and protein expression in the DLPFC of subjects with schizophrenia: Implications for cognitive deficits. Schizophr Bull 33: 259.
Eggan SM, Lewis DA (2007). Immunocytochemical distribution of the cannabinoid CB1 receptor in the primate neocortex: a regional and laminar analysis. Cereb Cortex 17: 175–191.
Elvevag B, Goldberg TE (2000). Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol 14: 1–21.
Erickson SL, Lewis DA (2002). Postnatal development of parvalbumin- and GABA transporter-immunoreactive axon terminals in monkey prefrontal cortex. J Comp Neurol 448: 186–202.
Erickson SL, Lewis DA (2004). Cortical connections of the lateral mediodorsal thalamus in cynomolgus monkeys. J Comp Neurol 473: 107–127.
Feinberg I (1982). Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatry Res 17: 319–334.
Fergusson DM, Poulton R, Smith PF, Boden JM (2006). Cannabis and psychosis. BMJ 332: 172–175.
Frankle WG, Laruelle M, Haber SN (2006). Prefrontal cortical projections to the midbrain in primates: evidence for a sparse connection. Neuropsychopharmacology 31: 1627–1636.
Fritschy J-M, Paysan J, Enna A, Mohler H (1994). Switch in the expression of rat GABAA-receptor subtypes during postnatal development: an immunohistochemical study. J Neurosci 14: 5302–5324.
Gabbott PLA, Bacon SJ (1996). Local circuit neurons in the medial prefrontal cortex (areas 24a,b,c, 25 and 32) in the monkey: II. Quantitative areal and laminar distributions. J Comp Neurol 364: 609–636.
Gabriel SM, Davidson M, Haroutunian V, Powchik P, Bierer LM, Purohit DP et al (1996). Neuropeptide deficits in schizophrenia vs Alzheimer's disease cerebral cortex. Biol Psychiatry 39: 82–91.
Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM et al (1998). Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry 65: 446–453.
Genoud C, Knott GW, Sakata K, Lu B, Welker E (2004). Altered synapse formation in the adult somatosensory cortex of brain-derived neurotrophic factor heterozygote mice. J Neurosci 24: 2394–2400.
Giedd JN (1999). Brain development during childhood and adolescence: a longitudinal MRI study. Nat Neurosci 2: 861–863.
Giguere M, Goldman-Rakic PS (1988). Mediodorsal nucleus: areal, laminar, and tangential distribution of afferents and efferents in the frontal lobe of rhesus monkeys. J Comp Neurol 277: 195–213.
Gilbert AR, Rosenberg DR, Harenski K, Spencer S, Sweeney JA, Keshavan MS (2001). Thalamic volumes in patients with first-episode schizophrenia. Am J Psychiatry 158: 618–624.
Gilbert CD, Kelly JP (1975). The projections of cells in different layers of the cat's visual cortex. J Comp Neurol 63: 81–106.
Glantz LA, Lewis DA (1997). Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia: regional and diagnostic specificity. Arch Gen Psychiatry 54: 943–952.
Glantz LA, Lewis DA (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57: 65–73.
Gogtay N, Giedd JN, Lusk L, Hayashi KM, Greenstein D, Vaituzis AC et al (2004). Dynamic mapping of human cortical development during childhood through early adulthood. Proc Natl Acad Sci USA 101: 8174–8179.
Gold JM (2004). Cognitive deficits as treatment targets in schizophrenia. Schizophr Res 72: 21–28.
Goldman-Rakic PS (1995). Cellular basis of working memory. Neuron 14: 477–485. This excellent review summarizes the cellular mechanisms underlying persistent activity of primate DLPFC pyramidal neurons during working memory tasks, with an emphasis on the recurrent excitatory interactions between DLPFC pyramidal cells.
Goldman-Rakic PS, Brown RM (1982). Postnatal development of monoamine content and synthesis in the cerebral cortex of rhesus monkeys. Brain Res 256: 339–349.
Goldman-Rakic PS, Lidow MS, Gallagher DW (1990). Overlap of dopaminergic, adrenergic, and serotoninergic receptors and complementarity of their subtypes in primate prefrontal cortex. J Neurosci 10: 2125–2138.
González-Albo MC, Elston GN, DeFelipe J (2001). The human temporal cortex: characterization of neurons expressing nitric oxide synthase, neuropeptides and calcium-binding proteins, and their glutamate receptor subunit profiles. Cereb Cortex 11: 1170–1181.
Gonzalez-Burgos G, Hashimoto T, Lewis DA (2007a). Inhibition and timing in cortical neural circuits. Am J Psychiatry 164: 12.
Gonzalez-Burgos G, Kroener S, Seamans JK, Lewis DA, Barrionuevo G (2005). Dopaminergic modulation of short-term synaptic plasticity in fast-spiking interneurons of primate dorsolateral prefrontal cortex. J Neurophysiol 94: 4168–4177.
Gonzalez-Burgos G, Kroener S, Zaitsev AV, Povysheva NV, Krimer LS, Barrionuevo G et al (2007b). Functional maturation of excitatory synapses in layer 3 pyramidal neurons during postnatal development of the primate prefrontal cortex. Cereb Cortex (in press; E-pub ahead of print).
González-Burgos G, Kroner S, Krimer LS, Seamans JK, Urban NN, Henze DA et al (2002). Dopamine modulation of neuronal function in the monkey prefrontal cortex. Physiol Behav 77: 537–543.
Grace AA, Floresco SB, Goto Y, Lodge DJ (2007). Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci 30: 220–227.
Green MF (1996). What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry 153: 321–330.
Guidotti A, Auta J, Davis JM, Gerevini VD, Dwivedi Y, Grayson DR et al (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder. Arch Gen Psychiatry 57: 1061–1069.
Guillery RW, Sherman SM (2002). Thalamic relay functions and their role in corticocortical communication: generalizations from the visual system. Neuron 33: 163–175.
Guo N, Hwang DR, Lo ES, Huang YY, Laruelle M, Abi-Dargham A (2003). Dopamine depletion and in vivo binding of PET D1 receptor radioligands: implications for imaging studies in schizophrenia. Neuropsychopharmacology 28: 1703–1711.
Hajos N, Katona I, Naiem SS, Mackie K, Ledent C, Mody I et al (2000). Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci 12: 3239–3249.
Hanada S, Mita T, Nishino N, Tanaka C (1987). [3H]muscimol binding sites increased in autopsied brains of chronic schizophrenics. Life Sci 40: 239–266.
Harrison PJ, Weinberger DR (2005). Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 10: 40–68.
Hashimoto T, Arion D, Volk DW, Mirnics K, Lewis D (2007a). Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry (E-pub ahead of print).
Hashimoto T, Bazmi HH, Mirnics K, Wu Q, Sampson AR, Lewis DA (2007b). Regional survey of GABA-related gene expression in the neocortex of subjects with schizophrenia. Schizophr Bull 33: 261.
Hashimoto T, Bergen SE, Nguyen QL, Xu B, Monteggia LM, Pierri JN et al (2005). Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J Neurosci 25: 372–383.
Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z et al (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 23: 6315–6326. This study provided direct evidence for cell type specificity in altered markers of GABA neurotransmission in schizophrenia.
Hayashi K, Ishikawa R, Ye LH, He XL, Takata K, Kohama K et al (1996). Modulatory role of drebrin on the cytoskeleton within dendritic spines in the rat cerebral cortex. J Neurosci 16: 7161–7170.
Hayes TL, Lewis DA (1996). Magnopyramidal neurons in the anterior motor speech region: dendritic features and interhemispheric comparisons. Arch Neurol 53: 1277–1283.
Hendry SHC, Jones EG, Emson PC (1984). Morphology, distribution, and synaptic relations of somatostatin- and neuropeptide Y-immunoreactive neurons in rat and monkey neocortex. J Neurosci 4: 2497–2517.
Henze DA, Gonzalez-Burgos GR, Urban NN, Lewis DA, Barrionuevo G (2000). Dopamine increases excitability of pyramidal neurons in primate prefrontal cortex. J Neurophysiol 84: 2799–2809.
Hill JJ, Hashimoto T, Lewis DA (2006). Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry 11: 557–566.
Hill JJ, Kolluri N, Hashimoto T, Wu Q, Sampson AR, Monteggia LM et al (2005). Analysis of pyramidal neuron morphology in an inducible knockout of brain-derived neurotrophic factor. Biol Psychiatry 57: 932–934.
Hirvonen J, Van Erp TG, Huttunen J, Aalto S, Nagren K, Huttunen M et al (2006). Brain dopamine d1 receptors in twins discordant for schizophrenia. Am J Psychiatry 163: 1747–1753.
Hof PR, Ungerleider LG, Webster MJ, Gattass R, Adams MM, Sailstad CA et al (1996). Neurofilament protein in differentially distributed in subpopulations of corticocortical projection neurons in the macaque monkey visual pathways. J Comp Neurol 376: 112–127.
Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K (2006). Experience-dependent and cell-type-specific spine growth in the neocortex. Nature 441: 979–983.
Howard MW, Rizzuto DS, Caplan JB, Madsen JR, Lisman J, Aschenbrenner-Scheibe R et al (2003). Gamma oscillations correlate with working memory load in humans. Cereb Cortex 13: 1369–1374. This intracranial recording study demonstrated that the power of γ band oscillations increases in proportion to working memory load in the human prefrontal cortex.
Huttenlocher PR, Dabholkar AS (1997). Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol 387: 167–178.
Jacobs B, Driscoll L, Schall M (1997). Life-span dendritic and spine changes in areas 10 and 18 of human cortex: a quantitative Golgi study. J Comp Neurol 386: 661–680.
Jones EG (1984). Laminar distribution of cortical efferent cells. In: Peters A, Jones EG (eds). Cerebral Cortex, vol. 1. Plenum Press: New York. pp 521–553.
Jourdain P, Fukunaga K, Muller D (2003). Calcium/calmodulin-dependent protein kinase II contributes to activity-dependent filopodia growth and spine formation. J Neurosci 23: 10645–10649.
Kalus P, Müller TJ, Zuschratter W, Senitz D (2000). The dendritic architecture of prefrontal pyramidal neurons in schizophrenic patients. NeuroReport 11: 3621–3625.
Karlsson P, Farde L, Halldin C, Sedvall G (2002). PET study of D1 dopamine receptor binding in neuroleptic-naive patients with schizophrenia. Am J Psychiatry 159: 761–767.
Karson CN, Mrak RE, Schluterman KO, Sturner WQ, Sheng JG, Griffin WST (1999). Alterations in synaptic proteins and their encoding mRNAs in prefrontal cortex in schizophenia: a possible neurochemical basis for ‘hypofrontality’. Mol Psychiatry 4: 39–45.
Kastner A, Anglade P, Bounaix C, Damier P, Javoy-Agid F, Bromet N et al (1994). Immunohistochemical study of catechol-O-methyltransferase in the human mesostriatal system. Neuroscience 62: 449–457.
Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K et al (1999). Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19: 4544–4558.
Katz LC, Shatz CJ (1996). Synaptic activity and the construction of cortical circuits. Science 274: 1133–1138.
Kawaguchi Y, Kondo S (2002). Parvalbumin, somatostatin and cholecystokinin as chemical markers for specific GABAergic interneuron types in the rat frontal cortex. J Neurocytol 31: 277–287. This provides an excellent review of the characteristic features of different classes of cortical GABA neurons.
Kawaguchi Y, Kubota Y (1993). Correlation of physiological subgroupings of nonpyramidal cells with parvalbumin- and calbindinD28k-immunoreactive neurons in layer V of rat frontal cortex. J Neurophysiol 70: 387–396.
Kawaguchi Y, Kubota Y (1996). Physiological and morphological identification of somatostatin- or vasoactive intestinal polypeptide-containing cells among GABAergic cell subtypes in rat frontal cortex. J Neurosci 16: 2701–2715.
Keshavan MS, Anderson S, Pettegrew JW (1994). Is schizophrenia due to excessive synaptic pruning in the prefrontal cortex? The Feinberg hypothesis revisited. J Psychiatry Res 28: 239–265.
Kholodilov N, Yarygina O, Oo TF, Zhang H, Sulzer D, Dauer W et al (2004). Regulation of the development of mesencephalic dopaminergic systems by the selective expression of glial cell line-derived neurotrophic factor in their targets. J Neurosci 24: 3136–3146.
Klausberger T, Magill PJ, Marton LF, Roberts JDB, Cobden PM, Buzsaki G et al (2003). Brain-state- and cell-type specific firing of hippocampal interneurons in vivo. Nature 421: 844–848. This study demonstrated for the first time in vivo that GABA neurons play specialized roles in regulating the synchronous activity of neuronal networks.
Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K (2006). Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci 9: 1117–1124.
Kolluri N, Sun Z, Sampson AR, Lewis DA (2005). Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry 162: 1200–1202.
Konradi C, Heckers S (2003). Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther 97: 153–179.
Kreczmanski P, Heinsen H, Mantua V, Woltersdorf F, Masson T, Ulfig N et al (2007). Volume, neuron density and total neuron number in five subcortical regions in schizophrenia. Brain 130: 678–692.
Krimer LS, Zaitsev AV, Czanner G, Kroner S, Gonzalez-Burgos G, Povysheva NV et al (2005). Cluster analysis-based physiological classification and morphological properties of inhibitory neurons in layers 2–3 of monkey dorsolateral prefrontal cortex. J Neurophysiol 94: 3009–3022.
Kristiansen LV, Beneyto M, Haroutunian V, Meador-Woodruff JH (2006). Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol Psychiatry 11: 737–747.
Kristiansen LV, Huerta I, Beneyto M, Meador-Woodruff JH (2007). NMDA receptors and schizophrenia. Curr Opin Pharmacol 7: 48–55. This informative review summarizes recent findings on the alterations of glutamate receptor proteins and mRNAs in the brain of subjects with schizophrenia.
Kroner S, Krimer LS, Lewis DA, Barrionuevo G (2007). Dopamine increases inhibition in the monkey dorsolateral prefrontal cortex through cell type-specific modulation of interneurons. Cereb Cortex 17: 1020–1032.
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD et al (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51: 199–214.
Kubota Y, Hattori R, Yui Y (1994). Three distinct subpopulations of GABAergic neurons in rat frontal agranular cortex. Brain Res 649: 159–173.
Lambe EK, Krimer LS, Goldman-Rakic PS (2000). Differential postnatal development of catecholamine and serotonin inputs to identified neurons in prefrontal cortex of rhesus monkey. J Neurosci 20: 8780–8788.
Lavoie AM, Tingey JJ, Harrison NL, Pritchett DB, Twyman RE (1997). Activation and deactivation rates of recombinant GABAA receptor channels are dependent on α-subunit isoform. Biophys J 73: 2518–2526.
Law AJ, Harrison PJ (2003). The distribution and morphology of prefrontal cortex pyramidal neurons identified using anti-neurofilament antibodies SMI32, N200 and FNP7. Normative data and a comparison in subjects with schizophrenia, bipolar disorder or major depression. J Psychiatr Res 37: 487–499.
Le Be JV, Markram H (2006). Spontaneous and evoked synaptic rewiring in the neonatal neocortex. Proc Natl Acad Sci USA 103: 13214–13219.
Levitt JB, Lewis DA, Yoshioka T, Lund JS (1993). Topography of pyramidal neuron intrinsic connections in macaque monkey prefrontal cortex (areas 9 & 46). J Comp Neurol 338: 360–376.
Lewis DA (2004). Structure of the human prefrontal cortex. Am J Psychiatry 161: 1366.
Lewis DA, Cruz DA, Melchitzky DS, Pierri JN (2001a). Lamina-specific deficits in parvalbumin-immunoreactive varicosities in the prefrontal cortex of subjects with schizophrenia: Evidence for fewer projections from the thalamus. Am J Psychiatry 158: 1411–1422.
Lewis DA, Gonzalez-Burgos G (2006). Pathophysiologically based treatment interventions in schizophrenia. Nat Med 12: 1016–1022.
Lewis DA, Hashimoto T, Volk DW (2005). Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 6: 312–324.
Lewis DA, Levitt P (2002). Schizophrenia as a disorder of neurodevelopment. Ann Rev Neurosci 25: 409–432.
Lewis DA, Melchitzky DS, Sesack SR, Whitehead RE, Auh S, Sampson AR (2001b). Dopamine transporter immunoreactivity in monkey cerebral cortex: regional, laminar and ultrastructural localization. J Comp Neurol 432: 119–138.
Lewis DA, Morrison JH (1989). The noradrenergic innervation of monkey prefrontal cortex: a dopamine-beta-hydroxylase immunohistochemical study. J Comp Neurol 282: 317–330.
Lidow MS, Goldman-Rakic PS (1994). A common action of clozapine, haloperidol, and remoxipride on D1- and D2-dopaminergic receptors in the primate cerebral cortex. Proc Natl Acad Sci USA 91: 4353–4356.
Lidow MS, Goldman-Rakic PS (1997). Differential regulation of D2 and D4 dopamine receptor mRNAs in the primate cerebral cortex vs neostriatum: effects of chronic treatment with typical and atypical antipsychotic drugs. J Pharmacol Exp Ther 283: 939–946.
Loup F, Weinmann O, Yonekawa Y, Aguzzi A, Wieser H-G, Fritschy J-M (1998). A highly sensitive immunofluorescence procedure for analyzing the subcellular distribution of GABAA receptor subunits in the human brain. J Histochem Cytochem 46: 1129–1139.
Luna B, Garver KE, Urban TA, Lazar NA, Sweeney JA (2004). Maturation of cognitive processes from late childhood to adulthood. Child Dev 75: 1357–1372.
Lund JS, Lewis DA (1993). Local circuit neurons of developing and mature macaque prefrontal cortex: Golgi and immunocytochemical characteristics. J Comp Neurol 328: 282–312.
Lund JS, Lund RD, Hendrickson AE, Bunt AH, Fuchs AF (1975). The origin of efferent pathways from the primary visual cortex, area 17, of the macaque monkey as shown by retrograde transport of horseradish peroxidase. J Comp Neurol 164: 287–304.
Lundstrom K, Tenhunen J, Tilgmann C, Karhunen T, Panula P, Ulmanen I (1995). Cloning, expression and structure of catechol-O-methyltransferase. Biochim Biophys Acta 1251: 1–10.
Luo L (2000). Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci 1: 173–180.
Ma XM, Huang J, Wang Y, Eipper BA, Mains RE (2003). Kalirin, a multifunctional Rho guanine nucleotide exchange factor, is necessary for maintenance of hippocampal pyramidal neuron dendrites and dendritic spines. J Neurosci 23: 10593–10603.
Ma Y, Hu H, Berrebi AS, Mathers PH, Agmon A (2006). Distinct subtypes of somatostatin-containing neocortical interneurons revealed in transgenic mice. J Neurosci 26: 5069–5082.
MacDonald III AW, Carter CS, Kerns JG, Ursu S, Barch DM, Holmes AJ et al (2005). Specificity of prefrontal dysfunction and context processing deficits to schizophrenia in never-medicated patients with first-episode psychosis. Am J Psychiatry 162: 475–484.
Maldonado-Aviles JG, Wu Q, Sampson AR, Lewis DA (2006). Somal size of immunolabeled pyramidal cells in the prefrontal cortex of subjects with schizophrenia. Biol Psychiatry 60: 226–234.
Mannisto PT, Kaakkola S (1999). Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev 51: 593–628.
Manoach DS, Press DZ, Thangaraj V, Searl MM, Goff DC, Halpern E et al (1999). Schizophrenic subjects activate dorsolateral prefrontal cortex during a working memory task, as measured by fMRI. Biol Psychiatry 45: 1128–1137.
Matsumoto M, Weickert CS, Beltaifa S, Kolachana B, Chen J, Hyde TM et al (2003). Catechol-O-methyltransferase (COMT) mRNA expression in the dorsolateral prefrontal cortex of patients with schizophrenia. Neuropsychopharmacology 28: 1521–1530.
Matsuzaki M (2007). Factors critical for the plasticity of dendritic spines and memory storage. Neurosci Res 57: 1–9.
Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4: 1086–1092.
McAllister AK, Katz LC, Lo DC (1996). Neurotrophin regulation of cortical dendritic growth requires activity. Neuron 17: 1057–1064.
McAllister AK, Lo DC, Katz LC (1995). Neurotrophins regulate dendritic growth and developing visual cortex. Neuron 15: 791–803.
Melchitzky DS, Eggan SM, Lewis DA (2005). Synaptic targets of calretinin-containing axon terminals in macaque monkey prefrontal cortex. Neuroscience 130: 185–195.
Melchitzky DS, González-Burgos G, Barrionuevo G, Lewis DA (2001). Synaptic targets of the intrinsic axon collaterals of supragranular pyramidal neurons in monkey prefrontal cortex. J Comp Neurol 430: 209–221.
Melchitzky DS, Lewis DA (2003). Pyramidal neuron local axon terminals in monkey prefrontal cortex: Differential targeting of subclasses of GABA neurons. Cereb Cortex 13: 452–460.
Melchitzky DS, Sesack SR, Lewis DA (1999). Parvalbumin-immunoreactive axon terminals in macaque monkey and human prefrontal cortex: laminar, regional and target specificity of Type I and Type II synapses. J Comp Neurol 408: 11–22.
Melchitzky DS, Sesack SR, Pucak ML, Lewis DA (1998). Synaptic targets of pyramidal neurons providing intrinsic horizontal connections in monkey prefrontal cortex. J Comp Neurol 390: 211–224.
Meyer-Lindenberg A, Nichols T, Callicott JH, Ding J, Kolachana B, Buckholtz J et al (2006). Impact of complex genetic variation in COMT on human brain function. Mol Psychiatry 11: 797, 867–877.
Mirnics K, Middleton FA, Lewis DA, Levitt P (2001). Analysis of complex brain disorders with gene expression microarrays: Schizophrenia as a disease of the synapse. Trends Neurosci 24: 479–486.
Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P (2000). Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 28: 53–67.
Missale C, Nash SR, Robinson SW, Jaber M, Caron MG (1998). Dopamine receptors: from structure to function. Physiol Rev 78: 189–225.
Moghaddam B (2003). Bringing order to the glutamate chaos in schizophrenia. Neuron 40: 881–884.
Muller M, Felmy F, Schwaller B, Schneggenburger R (2007). Parvalbumin is a mobile presynaptic Ca2+ buffer in the calyx of held that accelerates the decay of Ca2+ and short-term facilitation. J Neurosci 27: 2261–2271.
Muly EC, Szigeti K, Goldman-Rakic PS (1998). D1 receptor in interneurons of macaque prefrontal cortex: distribution and subcellular location. J Neurosci 18: 10553–10565.
Nakayama AY, Harms MB, Luo L (2000). Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci 20: 5329–5338.
Negishi M, Katoh H (2005). Rho family GTPases and dendrite plasticity. Neuroscientist 11: 187–191.
Neve KA, Seamans JK, Trantham-Davidson H (2004). Dopamine receptor signaling. J Recept Signal Transduct Res 24: 165–205.
Nobes CD, Hall A (1995). Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81: 53–62.
Novak G, Tallerico T (2006). Nogo A, B and C expression in schizophrenia, depression and bipolar frontal cortex, and correlation of Nogo expression with CAA/TATC polymorphism in 3′-UTR. Brain Res 1120: 161–171.
Nusser Z, Sieghart W, Benke D, Fritschy J-M, Somogyi P (1996). Differential synaptic localization of two major γ-aminobutyric acid type A receptor α subunits on hippocampal pyramidal cells. Proc Natl Acad Sci USA 93: 11939–11944.
Ohnuma T, Augood SJ, Arai H, McKenna PJ, Emson PC (1999). Measurement of GABAergic parameters in the prefrontal cortex in schizophrenia: focus on GABA content, GABAA receptor α-1 subunit messenger RNA and human GABA transporter-1 (HGAT-1) messenger RNA expression. Neuroscience 93: 441–448.
Okubo Y, Suhara T, Suzuki K, Kobayashi K, Inoue O, Terasaki O et al (1997). Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 385: 634–636.
Olney JW, Farber NB (1995). Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry 52: 998–1007.
Overstreet LS, Westbrook GL (2003). Synapse density regulates independence at unitary inhibitory synapses. J Neurosci 23: 2618–2626.
Pakkenberg B (1990). Pronounced reduction of total neuron number in mediodorsal thalamic nucleus and nucleus accumbens in schizophrenics. Arch Gen Psychiatry 47: 1023–1028.
Paspalas CD, Goldman-Rakic PS (2004). Microdomains for dopamine volume neurotransmission in primate prefrontal cortex. J Neurosci 24: 5292–5300.
Paspalas CD, Goldman-Rakic PS (2005). Presynaptic D1 dopamine receptors in primate prefrontal cortex: target-specific expression in the glutamatergic synapse. J Neurosci 25: 1260–1267.
Penzes P, Johnson RC, Alam MR, Kambampati V, Mains RE, Eipper BA (2000). An isoform of kalirin, a brain-specific GDP/GTP exchange factor, is enriched in the postsynaptic density fraction. J Biol Chem 275: 6395–6403.
Penzes P, Johnson RC, Sattler R, Zhang X, Huganir RL, Kambampati V et al (2001). The neuronal Rho-GEF Kalirin-7 interacts with PDZ domain-containing proteins and regulates dendritic morphogenesis. Neuron 29: 229–242.
Perez-Costas E, Melendez-Ferro M, Gao XM, Conley R, Roberts RC (2007). Decreased expression of tyrosine hydroxylase in the substantia nigra of schizophrenia brains. Schizophr Bull 33: 266.
Perrone-Bizzozero NI, Sower AC, Bird ED, Benowitz LI, Ivins KJ, Neve RL (1996). Levels of the growth-associated protein GAP-43 are selectively increased in association cortices in schizophrenia. Proc Natl Acad Sci USA 93: 14182–14187.
Peters A, Proskauer CC, Ribak CE (1982). Chandelier neurons in rat visual cortex. J Comp Neurol 206: 397–416.
Pierri JN, Volk CL, Auh S, Sampson A, Lewis DA (2003). Somal size of prefrontal cortical pyramidal neurons in schizophrenia: differential effects across neuronal subpopulations. Biol Psychiatry 54: 111–120.
Pierri JN, Volk CLE, Auh S, Sampson A, Lewis DA (2001). Decreased somal size of deep layer 3 pyramidal neurons in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry 58: 466–473.
Popken GJ, Bunney Jr WE, Potkin SG, Jones EG (2000). Subnucleus-specific loss of neurons in medial thalamus of schizophrenics. Proc Natl Acad Sci USA 97: 9276–9280.
Pouille F, Scanziani M (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293: 1159–1163.
Pucak ML, Levitt JB, Lund JS, Lewis DA (1996). Patterns of intrinsic and associational circuitry in monkey prefrontal cortex. J Comp Neurol 376: 614–630.
Rajkowska G, Selemon LD, Goldman-Rakic PS (1998). Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch Gen Psychiatry 55: 215–224. This study provided quantitative evidence of alterations in pyramidal cell morphology in schizophrenia.
Ramakers GJA (2002). Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci 25: 191–199.
Rao SG, Williams GV, Goldman-Rakic PS (2000). Destruction and creation of spatial tuning by disinhibition: GABAA blockade of prefrontal cortical neurons engaged by working memory. J Neurosci 20: 485–494.
Rosenberg DR, Lewis DA (1995). Postnatal maturation of the dopaminergic innervation of monkey prefrontal and motor cortices: a tyrosine hydroxylase immunohistochemical analysis. J Comp Neurol 358: 383–400.
Rotaru DC, Lewis DA, Gonzalez-Burgos G (2007). Dopamine D1 receptor activation regulates sodium channel-dependent EPSP amplification in rat prefrontal cortex pyramidal neurons. J of Physiology 581: 981–1000.
Rybakowski JK, Borkowska A, Czerski PM, Dmitrzak-Weglarz M, Skibinska M, Kapelski P et al (2006). Performance on the Wisconsin Card Sorting Test in schizophrenia and genes of dopaminergic inactivation (COMT, DAT, NET). Psychiatry Res 143: 13–19.
Sawaguchi T (2001). The effects of dopamine and its antagonists on directional delay-period activity of prefrontal neurons in monkeys during an oculomotor delayed-response task. Neurosci Res 41: 115–128.
Sawaguchi T, Goldman-Rakic PS (1991). D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science 251: 947–950. This study demonstrated for the first time that activation of D1 receptors in the prefrontal cortex is strictly necessary for working memory performance.
Sawaguchi T, Goldman-Rakic PS (1994). The role of D1-dopamine receptor in working memory: local injections of dopamine antagonists into the prefrontal cortex of rhesus monkeys performing an oculomotor delayed-response task. J Neurophysiol 71: 515–528.
Sawaguchi T, Matsumura M, Kubota K (1989). Delayed response deficits produced by local injection of bicuculline into the dorsolateral prefrontal cortex in Japanese macaque monkeys. Exp Brain Res 75: 457–469.
Schneggenburger R, Sakaba T, Neher E (2002). Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci 25: 206–212.
Scott EK, Reuter JE, Luo L (2003). Small GTPase Cdc42 is required for multiple aspects of dendritic morphogenesis. J Neurosci 23: 3118–3123.
Seamans JK, Yang CR (2004). The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol 74: 1–58. This comprehensive review article provides an excellent summary of the cellular mechanisms of dopamine actions in prefrontal cortical circuits.
Selemon LD, Goldman-Rakic PS (1999). The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol Psychiatry 45: 17–25.
Selemon LD, Rajkowska G, Goldman-Rakic PS (1995). Abnormally high neuronal density in the schizophrenic cortex: a morphometric analysis of prefrontal area 9 and occipital area 17. Arch Gen Psychiatry 52: 805–818.
Sesack SR, Carr DB (2002). Selective prefrontal cortex inputs to dopamine cells: implications for schizophrenia. Physiol Behav 77: 513–517.
Sesack SR, Hawrylak VA, Melchitzky DS, Lewis DA (1998). Dopamine innervation of a subclass of local circuit neurons in monkey prefrontal cortex: ultrastructural analysis of tyrosine hydroxylase and parvalbumin immunoreactive structures. Cereb Cortex 8: 614–622.
Simpson MDC, Slater P, Deakin JFW, Royston MC, Skan WJ (1989). Reduced GABA uptake sites in the temporal lobe in schizophrenia. Neurosci Lett 107: 211–215.
Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS (1994). D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. Proc Natl Acad Sci USA 91: 5720–5724.
Soloway AS, Pucak ML, Melchitzky DS, Lewis DA (2002). Dendritic morphology of callosal and ipsilateral projection neurons in monkey prefrontal cortex. Neuroscience 109: 461–471.
Spencer KM, Nestor PG, Perlmutter R, Niznikiewicz MA, Klump MC, Frumin M et al (2004). Neural synchrony indexes disordered perception and cognition in schizophrenia. Proc Natl Acad Sci USA 101: 17288–17293.
Stein V, Nicol RA (2003). GABA generates excitement. Neuron 37: 375–378.
Stettler DD, Yamahachi H, Li W, Denk W, Gilbert CD (2006). Axons and synaptic boutons are highly dynamic in adult visual cortex. Neuron 49: 877–887.
Straub RE, Lipska BK, Egan MF, Goldberg TE, Callicot JH, Mayhew MB et al (2007). Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol Psychiatry (E-pub ahead of print).
Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C et al (2006). Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci 9: 99–107.
Sweet RA, Bergen SE, Sun Z, Sampson AR, Pierri JN, Lewis DA (2004). Pyramidal cell size reduction in schizophrenia: evidence for involvement of auditory feedforward circuits. Biol Psychiatry 55: 1128–1137.
Sweet RA, Pierri JN, Auh S, Sampson AR, Lewis DA (2003). Reduced pyramidal cell somal volume in auditory association cortex of subjects with schizophrenia. Neuropsychopharmacology 28: 599–609.
Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G (2006). Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311: 233–235.
Tada T, Sheng M (2006). Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol 16: 95–101.
Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T (2003). Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J Neurosci 23: 6586–6595.
Tan HY, Choo WC, Fones CS, Chee MW (2005). fMRI study of maintenance and manipulation processes within working memory in first-episode schizophrenia. Am J Psychiatry 162: 1849–1858.
Tassin JP, Studler JM, Hervé D, Blanc G, Glowinski J (1986). Contribution of noradrenergic neurons to the regulation of dopaminergic [D(1)] receptor denervation supersensitivity in rat prefrontal cortex. J Neurochem 46: 243–248.
Thune JJ, Uylings HBM, Pakkenberg B (2001). No deficit in total number of neurons in the prefrontal cortex in schizophrenics. J Psychiatr Res 35: 15–21.
Torrey EF, Barci BM, Webster MJ, Bartko JJ, Meador-Woodruff JH, Knable MB (2005). Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol Psychiatry 57: 252–260.
Tunbridge E, Burnet PW, Sodhi MS, Harrison PJ (2004a). Catechol-O-methyltransferase (COMT) and proline dehydrogenase (PRODH) mRNAs in the dorsolateral prefrontal cortex in schizophrenia, bipolar disorder, and major depression. Synapse 51: 112–118.
Tunbridge EM, Bannerman DM, Sharp T, Harrison PJ (2004b). Catechol-O-methyltransferase inhibition improves set-shifting performance and elevates stimulated dopamine release in the rat prefrontal cortex. J Neurosci 24: 5331–5335.
Tunbridge EM, Weinberger DR, Harrison PJ (2006). A novel protein isoform of catechol-O-methyltransferase (COMT): brain expression analysis in schizophrenia and bipolar disorder and effect of Val158Met genotype. Mol Psychiatry 11: 116–117.
Vawter MP, Crook JM, Hyde TM, Kleinman JE, Weinberger DR, Becker KG et al (2002). Microarray analysis of gene expression in the prefrontal cortex in schizophrenia: a preliminary study. Schizophr Res 58: 11–20.
Verma A, Moghaddam B (1996). NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: modulation by dopamine. J Neurosci 16: 373–379.
Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF (2007). Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci 10: 376–384.
Virgo L, Humphries C, Mortimer A, Barnes T, Hirsch SR, de Belleroche J (1995). Cholecystokinin messenger RNA deficit in frontal and temporal cerebral cortex in schizophrenia. Biol Psychiatry 37: 694–701.
Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA (2000). Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry 57: 237–245.
Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA (2001). GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry 158: 256–265.
Volk DW, Pierri JN, Fritschy J-M, Auh S, Sampson AR, Lewis DA (2002). Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb Cortex 12: 1063–1070.
Vreugdenhil M, Jefferys JG, Celio MR, Schwaller B (2003). Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. J Neurophysiol 89: 1414–1422.
Waites CL, Craig AM, Garner CC (2005). Mechanisms of vertebrate synaptogenesis. Annu Rev Neurosci 28: 251–274.
Wang XJ (2006). Toward a prefrontal microcircuit model for cognitive deficits in schizophrenia. Pharmacopsychiatry 39 (Suppl 1): S80–S87.
Weickert CS, Hyde TM, Lipska BK, Herman MM, Weinberger DR, Kleinman JE (2003). Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry 8: 592–610.
Weickert CS, Ligons DL, Romanczyk T, Ungaro G, Hyde TM, Herman MM et al (2005). Reductions in neurotrophin receptor mRNAs in the prefrontal cortex of patients with schizophrenia. Mol Psychiatry 10: 637–650.
Weickert CS, Webster MJ, Gondipalli P, Rothmond D, Fatula RJ, Herman MM et al (2007). Postnatal alterations in dopaminergic markers in the human prefrontal cortex. Neuroscience 144: 1109–1119.
Weinberger DR (1987). Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 44: 660–669.
White EL (1989). Cortical Circuits. Boston-Basel: Birkhauser, 34 pp.
White FJ (1996). Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci 19: 405–436.
Whittington MA, Traub RD (2003). Interneuron diversity series: inhibitory interneurons and network oscillations in vitro. Trends Neurosci 26: 676–682.
Williams GV, Goldman-Rakic PS (1995). Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature 376: 572–575.
Williams HJ, Owen MJ, O'Donovan MC (2007). Is COMT a susceptibility gene for schizophrenia? Schizophr Bull 33: 635–641.
Williams SM, Goldman-Rakic PS, Leranth C (1992). The synaptology of parvalbumin-immunoreactive neurons in primate prefrontal cortex. J Comp Neurol 320: 353–369.
Woo T-U, Miller JL, Lewis DA (1997a). Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am J Psychiatry 154: 1013–1015.
Woo T-U, Pucak ML, Kye CH, Matus CV, Lewis DA (1997b). Peripubertal refinement of the intrinsic and associational circuitry in monkey prefrontal cortex. Neuroscience 80: 1149–1158.
Woo T-U, Walsh JP, Benes FM (2004). Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry 61: 649–657.
Woo T-U, Whitehead RE, Melchitzky DS, Lewis DA (1998). A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci USA 95: 5341–5346.
Yang CR, Seamans JK, Gorelova N (1999). Developing a neuronal model for the pathophysiology of schizophrenia based on the nature of electrophysiological actions of dopamine in the prefrontal cortex. Neuropsychopharmacology 21: 161–194.
Young KA, Holcomb LA, Yazdani U, Hicks PB, German DC (2004). Elevated neuron number in the limbic thalamus in major depression. Am J Psychiatry 161: 1270–1277.
Young KA, Manaye KF, Liang C-L, Hicks PB, German DC (2000). Reduced number of mediodorsal and anterior thalamic neurons in schizophrenia. Biol Psychiatry 47: 944–953.
Zaitsev AV, Gonzalez-Burgos G, Povysheva NV, Kroner S, Lewis DA, Krimer LS (2005). Localization of calcium-binding proteins in physiologically and morphologically characterized interneurons of monkey dorsolateral prefrontal cortex. Cereb Cortex 15: 1178–1186.
Zhao X, Qin S, Shi Y, Zhang A, Zhang J, Bian L et al (2007). Systematic study of association of four GABAergic genes: glutamic acid decarboxylase 1 gene, glutamic acid decarboxylase 2 gene, GABA(B) receptor 1 gene and GABA(A) receptor subunit beta2 gene, with schizophrenia using a universal DNA microarray. Schizophr Res 93: 374–384.
Zhou Q, Homma KJ, Poo MM (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44: 749–757.
Zucker RS, Regehr WG (2002). Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405.
Zuo Y, Lin A, Chang P, Gan WB (2005). Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46: 181–189. This elegant imaging study used in vivo imaging techniques to demonstrate the remarkable stability of individual dendritic spines in the adult brain.
Acknowledgements
The works cited which we conducted were supported by NIH grants MH045156, MH051234, and MH043784, and by a NARSAD Young Investigator Award.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
David A Lewis currently receives research support from the BMS Foundation, Merck and Pfizer and in 2006–2007 served as a consultant to Pfizer, Roche, Sepracor, and Wyeth. Over the past three years, he also declares grant support from Lilly and has been a consultant to Aventis and Astra-Zeneca. Dr Guillermo Gonzalez-Burgos declares that, except for income received from my primary employer, no financial support or compensation has been received from any individual or corporate entity over the past three years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Rights and permissions
About this article
Cite this article
Lewis, D., González-Burgos, G. Neuroplasticity of Neocortical Circuits in Schizophrenia. Neuropsychopharmacol 33, 141–165 (2008). https://doi.org/10.1038/sj.npp.1301563
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301563
Keywords
This article is cited by
-
Synaptic plasticity and mental health: methods, challenges and opportunities
Neuropsychopharmacology (2023)
-
Neonatal phencyclidine as a model of sex-biased schizophrenia symptomatology in adolescent mice
Psychopharmacology (2023)
-
Roles of neuroligins in central nervous system development: focus on glial neuroligins and neuron neuroligins
Journal of Translational Medicine (2022)
-
Defective AMPA-mediated synaptic transmission and morphology in human neurons with hemizygous SHANK3 deletion engrafted in mouse prefrontal cortex
Molecular Psychiatry (2021)
-
Opposite alterations of 5HT2A receptor brain density in subjects with schizophrenia: relevance of radiotracers pharmacological profile
Translational Psychiatry (2021)
