AKT/GSK3 signaling pathway and schizophrenia
- Advanced Technologies for Novel Therapeutics (ATNT), Newark, NJ, USA
Schizophrenia is a prevalent complex trait disorder manifested by severe neurocognitive dysfunctions and lifelong disability. During the past few years several studies have provided direct evidence for the involvement of different signaling pathways in schizophrenia. In this review, we mainly focus on AKT/GSK3 signaling pathway in schizophrenia. The original study on the involvement of this pathway in schizophrenia was published by Emamian et al. in 2004. This study reported convergent evidence for a decrease in AKT1 protein levels and levels of phosphorylation of GSK-3β in the peripheral lymphocytes and brains of individuals with schizophrenia; a significant association between schizophrenia and an AKT1 haplotype; and a greater sensitivity to the sensorimotor gating-disruptive effect of amphetamine, conferred by AKT1 deficiency. It also showed that haloperidol can induce a stepwise increase in regulatory phosphorylation of AKT1 in the brains of treated mice that could compensate for the impaired function of this signaling pathway in schizophrenia. Following this study, several independent studies were published that not only confirmed the association of this signaling pathway with schizophrenia across different populations, but also shed light on the mechanisms by which AKT/GSK3 pathway may contribute to the development of this complex disorder. In this review, following an introduction on the role of AKT in human diseases and its functions in neuronal and non-neuronal cells, a review on the results of studies published on AKT/GSK3 signaling pathway in schizophrenia after the original 2004 paper will be provided. A brief review on other signaling pathways involved in schizophrenia and the possible connections with AKT/GSK3 signaling pathway will be discussed. Moreover, some possible molecular mechanisms acting through this pathway will be discussed besides the mechanisms by which they may contribute to the pathogenesis of schizophrenia. Finally, different transcription factors related to schizophrenia will be reviewed to see how hypo-activity of AKT signaling pathway may impact such transcriptional mechanisms.
Introduction
Schizophrenia is a severe mental disorder with a lifetime prevalence of 1% throughout the world population (Stilo and Murray, 2010). This highly prevalent mental disorder is typically associated with progressive functional decline, lifelong disability and tremendous suffering (Sadock et al., 2009). Unlike other neuropsychiatric disorders such as Parkinson's disease, no single anatomical abnormality is consistently observed in schizophrenia, nor there are any biochemical tests that can confirm the clinical diagnosis. Case identification usually begins with the observation of positive symptoms such as delusional ideas, hallucinations, disordered thinking, often with a later emergence of negative symptoms, including low levels of emotional arousal, mental activity and social drive (Sadock et al., 2009). Furthermore, there is increasing recognition of several cognitive impairments in this disorder, particularly in attention, memory, and executive functions, which equally contribute to schizophrenia's disability. Although it is believed that multiple pathological processes can lead to schizophrenia, we have neither identified them nor linked them to the various clinical manifestations of the disorder (Sadock et al., 2009).
Several linkage and association studies, including a few genome-wide association studies (GWAS), have been performed during the past decade to identify genetic susceptibilities to the disease. A few review articles have nicely reviewed recent association studies on large schizophrenia cohorts (Tiwari et al., 2010). Such studies identified and replicated further loci in well-powered cohorts (Tiwari et al., 2010). Moreover, over the past 4 years there has been a significant interest in human Copy Number Variants (CNVs). Several deletions that were recently identified in schizophrenia, such as 1q21.1; 2p16.3; 15q11.2; 15q13.3, have also been found in other neurodevelopmental disorders (Tiwari et al., 2010).
Despite identification of several genetic variations that have been repeatedly reproduced in several populations, many studies have been inconclusive. The limited success in detection of genetic factors has led scientists to come to a general consensus that the disease is not caused by the dysfunction of a specific molecule or pathway. Most likely schizophrenia is caused by the altered function or expression of many genes, which may individually contribute only to a small risk, but their collective dysfunctional effects interfere with the function of several biological pathways that eventually produce the clinical picture we know as schizophrenia (Williams et al., 2009).
AKT and Human Diseases
Deregulation of AKT signaling pathway is directly associated with some of the most prevalent and incurable human diseases (Blain and Massague, 2002; Liang et al., 2002; Shin et al., 2002; Viglietto et al., 2002; Chen et al., 2003; Emamian et al., 2003, 2004; Brazil et al., 2004; Colin et al., 2005; Griffin et al., 2005). It is now well established that the hyperactivity of AKT is part of the pathologic process in several types of the most common human malignancies (Brazil et al., 2004), including breast cancer, prostate cancer, lung cancer, gastrointestinal tumors, pancreatic cancer, hepatocellular carcinoma, thyroid cancer, and CNS malignancies (such as glioblastoma or gliomas). Moreover, impairment of AKT signaling pathway in a number of neurodegenerative brain disorders such as Alzheimer disease, Huntington disease, spinocerebellar ataxia type 1 (SCA1) and amyotrophic lateral sclerosis (ALS), have been reported during the past few years (Saudou et al., 1998; Chen et al., 2003; Emamian et al., 2003; Kaspar et al., 2003; Colin et al., 2005; Griffin et al., 2005).
In Alzheimer disease, evidence for an increased AKT activation and the subsequent increase in the phosphorylation of several AKT substrates in the brain tissue of patients has been reported (Griffin et al., 2005). In Huntington disease, in vitro studies have shown that huntingtin is a substrate of AKT, and the phosphorylation of huntingtin by AKT is crucial for mediating the neuroprotective effects of IGF-1. Furthermore, changes in AKT signaling in an animal model of Huntington disease, as well as in patients with Huntington disease, are also reported (Saudou et al., 1998; Colin et al., 2005). In SCA1, Chen et al. (2003) reports a regulatory role of AKT in the association of ataxin-1 with 14-3-3 through the phosphorylation of Ser-776 of ataxin-1, which modulates the neurotoxicity of ataxin-1 (Emamian et al., 2003). In ALS, IGF-1 stimulates the activity of AKT in the spinal cord and prolongs the lifespan of the mouse model of ALS, by increasing the survival of motor neurons (Kaspar et al., 2003).
Using a reverse protein-to-gene approach, we reported convergent evidence for the impairment of AKT/GSK3 signaling pathway in schizophrenia (Emamian et al., 2004), including a significant association between schizophrenia and an AKT1 haplotype in U.S. families (Caucasian of Northern European origin). The genetic association of AKT1 with schizophrenia was later confirmed in several other populations (Ikeda et al., 2004; Schwab et al., 2005; Bajestan et al., 2006). Taken together, it seems that the genetic association of AKT with schizophrenia is not limited to a specific genetic background and is reproducible in populations with different genetic backgrounds.
The main advantage of studies on signaling molecules in complex human disorders with unknown etiology, such as schizophrenia and autism, is their potential for discovering the abnormal function of proteins involved in the pathogenesis regardless the actual causative factor, whether it is genetic or environment or the interplay between them. Another important advantage of studies on signaling proteins such as AKT is their capacity for identification of sequence specific activation or inhibition sites on molecules involved in the disease process. Such specific sites could be ultimately used as selective targets for high throughput screening of small molecules or chemical entities, to discover novel therapeutics for several disorders. Therefore, despite the fact that schizophrenia is complex and extremely difficult to understand at the molecular level, dissecting the role of signaling molecules such as AKT or GSK3 in such a complex disorder has obvious therapeutic implications.
AKT in Non-Neuronal Cells
AKT (also known as PKB) is a relatively new member of the AGC kinase family. It was discovered in early 1990s as a major regulator of cell cycle (Brazil et al., 2004). After almost two decades, over 30,000 papers have been published (∼7000 in the past year) that show its major regulatory role in multiple aspects of cellular function. Figure 1 summarizes the molecular targets of AKT signaling pathway.
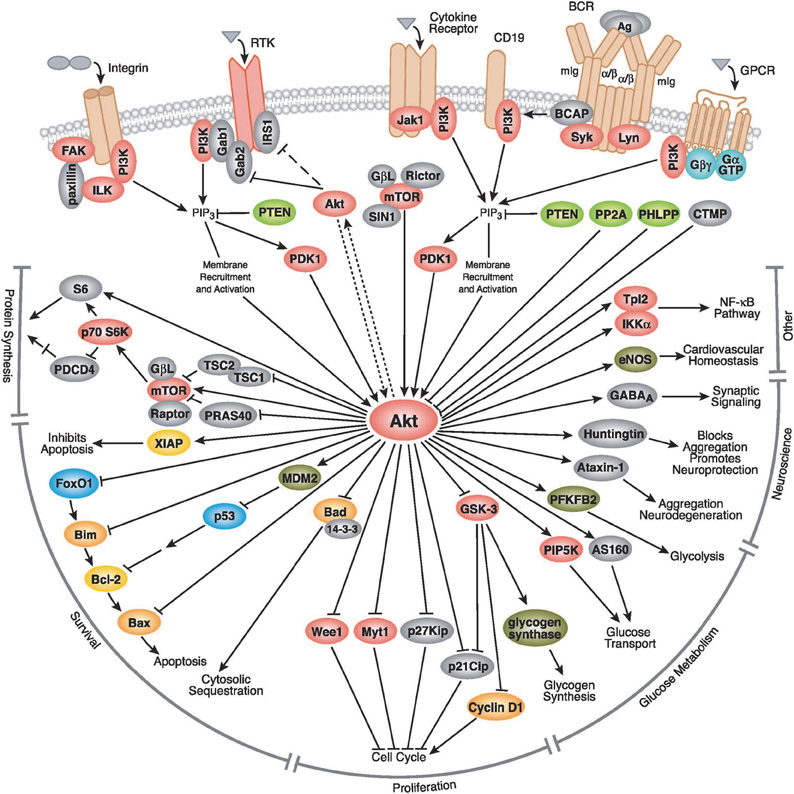
Figure 1. The AKT signaling pathway. AKT is activated by PI3K, which itself is activated by several upstream signaling pathways such as insulin receptors, receptor tyrosine kinases, G protein coupled receptors, cytokine receptors, etc. After activation, it targets several downstream molecules and change their activity by phosphorylation or complex formation. AKT is involved in cell proliferation, glucose metabolism, cell survival, cell cycle, protein synthesis, and in neuronal morphology and plasticity by regulation of several downstream molecules shown in this figure. AKT is involved in cell proliferation through interaction with a number of proteins involved in cell cycle, including Cyclin D1, p21Cip, p27Kip, Myt1, and Wee1. AKT also plays a crucial role in cell survival through interaction with Bad, MDM2 and the subsequent regulation of p53, FoxO1, Bcl-2, and Bax. AKT regulates glucose metabolism through regulation of GSK-3, PFKFB2, PIP5K, and AS160. It could also affect protein synthesis through interaction with S6 kinase, TSC1, and mTOR signaling. In more recent years, AKT's role in neurodegeneration was uncovered, mediated through phosphorylation of Huntingtin and Ataxin-1. AKT also has a role in synaptic plasticity, by phosphorylation of GABAA receptor. Pathway diagram reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com).
AKT has emerged as the focal point for many signal transduction pathways, regulating multiple cellular processes such as glucose metabolism, transcription, apoptosis, cell proliferation, angiogenesis, and cell motility (Figure 1, www.cellsignal.com, Kandel and Hay, 1999). Stimulation of its catalytic activity is triggered by PI3 kinase and results from the PtdIns(3,4,5)P3-dependent recruitment of AKT from the cytoplasm to the membrane, as well as the phosphorylation of two regulatory residues: Thr-308 and Ser-473. Phosphorylation of Thr-308, catalyzed by PDK-1, is required for the AKT activity, and this activity is augmented, ∼10-fold, by Ser-473 phosphorylation (Alessi et al., 1996; Brazil et al., 2004). Besides functioning as a kinase toward many substrates involved in these processes, AKT forms complexes with other proteins that are not substrates, but rather act as the modulators of the activity and function (Brazil et al., 2004).
AKT is highly expressed in different cell types (Brazil et al., 2004). AKT has three isoforms in mammalian cells: AKT1, AKT2, and AKT3, which have distinct roles in a variety of processes, including development and metabolism (Dummler and Hemmings, 2007). AKT1 is the most highly expressed isoform and is the isoform reported so far to be associated with schizophrenia (Emamian et al., 2004). AKT2 is mainly involved in regulation of metabolism primarily through insulin-regulated glucose homeostasis. AKT2 is expressed at a lower level than AKT1 and significant expression of AKT2 occurs in insulin-responsive tissues, such as skeletal muscle, liver, heart, kidney, and adipose tissue (Altomare et al., 1995; Chong et al., 2005). During development of central nervous system (CNS), the expression of AKT1 and AKT2 is increased during development, but is gradually decreased during postnatal development (Owada et al., 1997). In the adult brain, expression of AKT1 and AKT2 is initially weak. Unlike AKT1 and AKT2, AKT3 is only expressed in specific tissues, such as in the brain and testes, with lower expression evident in skeletal muscle, pancreas, heart, and kidney (Chong et al., 2005). Although AKT3 is expressed in the brain and has a role in postnatal brain development (Birnbaum, 2004; Tschopp et al., 2005; Gonzalez and McGraw, 2009), there is not that much know about its association with brain disorders. The focus of most studies related to schizophrenia is on AKT1, however, we cannot rule out the possible role of other isoforms of AKT in schizophrenia (Birnbaum, 2004; Freyberg et al., 2010).
AKT also plays a crucial role in cell survival (Brazil et al., 2004). Enhanced AKT activity can foster cell survival against several toxic insults such as matrix detachment (Rytomaa et al., 2000), DNA damage Henry et al., 2001; Kang et al., 2003a,b; Chong et al., 2005, anti-Fas antibody administration (Suhara et al., 2001), oxidative stress (Kang et al., 2003a,b; Chong et al., 2005), and transforming growth factor-ß application (Conery et al., 2004). Furthermore, many agents or growth factors can prevent apoptotic cellular injury through the activation of AKT. In the vascular system, angiopoietin-1 is an endothelium-specific ligand essential for embryonic vascular stabilization, branching, morphogenesis, and post-natal angiogenesis. Angiopoietin-1 also supports endothelial cell survival and prevents apoptosis through the activation of AKT (Papapetropoulos et al., 2000; Chong et al., 2005). In the cardiovascular system, myocardial protection by insulin during myocardial ischemia/reperfusion is eliminated by inhibition of PI3-K, suggesting that cardioprotection of insulin is mediated by AKT activation (Jonassen et al., 2001; Chong et al., 2005). Other trophic factors and cytokines, such as erythropoietin, may depend upon AKT to offer cellular protection (Chong et al., 2005).
AKT in Neurons
Signaling through protein phosphorylation is of importance for the normal development and function of the CNS. Protein phosphorylation is one of the most important mechanisms used in the nervous system for the regulation of the specificity and efficacy of neurotransmitter release from the presynaptic terminal in response to the nerve impulse (Greengard et al., 1993; Greengard, 2001).
AKT has a basic role in regulating neuronal cell size and survival (Franke, 2008; Niizuma et al., 2009; Freyberg et al., 2010). AKT is a critical survival factor that can modulate cellular pathways involved in apoptosis. Over-expression of AKT in cerebellar granule neurons prevents apoptosis during growth factor withdrawal (Dudek et al., 1997; Chong et al., 2005). However, expression of a dominant-negative AKT or inhibition of PI3-K attenuates neuronal cell survival normally supported by growth factors (Maiese and Chong, 2004; Chong et al., 2005). Several studies illustrated an important role for AKT for the survival of various neuronal cell types during cell injury. AKT promotes cell survival during free radical exposure in primary hippocampal neurons (Matsuzaki et al., 1999; Chong et al., 2005), neuronal cell lines (Kang et al., 2003a,b), and cerebral vascular endothelial cells (Chong et al., 2005).
During the past decade, there has been emerging evidence indicating several novel functions of AKT signaling pathway in the brain. In the CNS, AKT prevents injury-induced neuronal death and accelerates axonal regeneration (Namikawa et al., 2000). Moreover, it is shown that AKT is required for NGF-induced axon elongation and enhances axon branching (Markus et al., 2002). Intriguingly, AKT also plays a role in neuronal migration. Reelin, a large extracellular matrix protein responsible for positioning of cortical neurons, contributes to final neuron positioning in the mammalian brain by local modulation of AKT kinase activity (Beffert et al., 2002). The interesting study by Lin et al. (2001) provides a key piece in discovery of the roles played by AKT in the adult CNS. Their study shows AKT plays a crucial role in synaptic plasticity and memory formation. Another study by Mizuno et al. (2003) also shows that phosphorylation of BDNF receptor tyrosine kinase B (TrkB), PI3-K and its target AKT in the hippocampus increased in parallel with spatial memory formation (Mizuno et al., 2003). The study by Wang et al. (2003) uncovered a novel function of AKT in CNS. They discovered that AKT phosphorylates, both in vitro and in vivo, the type A GABA receptor, the principal receptor mediating fast inhibitory synaptic transmission, leading to increase in the number of GABA receptors on the plasma membrane surface, thereby increasing inhibitory fast synaptic transmission in neurons. Another suggested mechanism for AKT's role in synaptic plasticity and memory is through regulation of the mammalian target of rapamycin (mTOR). AKT is known to activate mTOR via phosphorylation and inhibition of the TSC2 subunit of the tuberous sclerosis complex (TSC). Interestingly, TSC2 heterozygous mice, in which the mTORC1 signaling pathways is constitutively active, exhibit impaired hippocampus-dependent memory that is reversible by rapamcyin treatment (Klann and Dever, 2004; Costa-Mattioli et al., 2009). Finally, the elegant study of Beaulieu et al. (2005) showed that AKT in complex with beta-arrestin 2 and PP2A mediates the dopaminergic neurotransmission and behavior.
Some other studies provided more interesting evidence showing the direct role of AKT is synaptic function and plasticity. AKT modulates synaptic plasticity and affects brain functions such as long-term potentiation, working memory, and fear conditioning (Lin et al., 2001; Lai et al., 2006; Sui et al., 2008; Niizuma et al., 2009; Freyberg et al., 2010). AKT also plays a role presynaptically through intracellular trafficking of biogenic amine transporters including the dopamine and norepinephrine transporters (Freyberg et al., 2010). Dopamine-mediated changes in AKT activity are thought to be mediated by postsynaptic dopamine D2 receptors (Beaulieu et al., 2007). In Drosophila, a study by Guo and Zhong (2006) shows the requirement of AKT for mediation of long-term depression. Moreover, in the mouse hippocampus, it is been shown that activation of the PI3-kinase and AKT is required for metabotropic glutamate receptor-dependent long-term depression (Hou and Klann, 2004). More evidence in support of this finding is provided by Lee et al. (2005), which shows stimulation of postsynaptic density-95 (PSD-95) protein translation by insulin via the PI3-kinase and AKT signaling pathway. Prior to this study, another report by Akama and McEwen (2003) shows that estrogen can also stimulate PSD-95 rapid protein synthesis via AKT signaling pathway.
AKT Signaling in Schizophrenia
Since the initial report in 2004 (Emamian et al., 2004), many other studies have been published supporting that impaired AKT signaling plays an important role in the pathogenesis of schizophrenia. This study and several subsequent ones uncovered association between AKT1 genetic variants and schizophrenia in case/control samples and family cohorts (Emamian et al., 2004; Ikeda et al., 2004; Schwab et al., 2005; Bajestan et al., 2006). The original study reported a significant association between schizophrenia and an AKT1 haplotype in U.S. families (Caucasian of Northern European origin). The genetic association of AKT1 with schizophrenia was later confirmed in European (Schwab et al., 2005), Japanese (Ikeda et al., 2004), Middle Eastern (Bajestan et al., 2006), Chinese (Xu et al., 2007) and Irish (Thiselton et al., 2008) populations. Taken together, despite the presence a negative report in a case-control Japanese population (Ohtsuki et al., 2004), it seems that the genetic association of AKT with schizophrenia is not limited to a specific genetic background and is reproducible in populations with different backgrounds.
Some studies have tried to study the correlations of the original SNPs reported in Emamian et al. (2004) with specific domains of human brain function. For example, Tan et al. (2008) examined the relationship of five original AKT1 SNPs to cognition in healthy subjects. They found that variants of the gene are associated with differences in specific domains of cognitive function including IQ, executive functioning, and processing speed. The executive functioning domain of brain is one of cognitive domains affected in schizophrenia (Sadock et al., 2009). Interestingly, Tan et al. also found that the single SNPs that is associated with reduced expression of AKT1 in peripheral lymphocytes, is also associated with brain volume reductions in caudate and right prefrontal cortex (Tan et al., 2008).
AKT1 protein level is significantly reduced in different tissues from patients with schizophrenia. Lymphocytes from individuals with schizophrenia expressed 68% less AKT1 relative to control subjects (Emamian et al., 2004). More importantly, AKT1 was reduced in the hippocampus and frontal cortex in postmortem brain samples from those with schizophrenia relative to comparison subjects. This decrease in protein level in the brain was specific to AKT1 isoform and AKT2 and AKT3 levels were unaffected (Emamian et al., 2004). Although some microarray studies failed to find changes in AKT1 mRNA levels in the brain tissue of schizophrenics (Horváth et al., 2011), a number of studies provided convergent evidence of a decrease in AKT1 mRNA, protein, and activity levels in the prefrontal cortex and hippocampus, as well as in peripheral blood of individuals with schizophrenia (Emamian et al., 2004; Zhao et al., 2006; Thiselton et al., 2008). The activity of major AKT1 targets such as GSK3 was also found to be altered in individuals with schizophrenia (Emamian et al., 2004). Several studies, described below, have since been published that uncover specific molecular mechanisms underlying the involvement of AKT signaling pathway in schizophrenia.
GSK-3 Signaling in Schizophrenia
Some studies have reported changes in the protein or activity levels of GSK-3β in different tissues obtained from schizophrenia samples (Koros and Dorner-Ciossek, 2007). Yang et al first reported decreased levels of GSK-3α proteins in lymphocytes of individuals with schizophrenia (Yang et al., 1995). We saw a statistically insignificant decrease in GSK-3β total protein levels in lymphocyte of patients with schizophrenia (Emamian et al., 2004). Further analysis of postmortem brains from patients with schizophrenia confirmed decreased phosphorylation levels and GSK-3β protein levels in the frontal cortex, as well as GSK-3β mRNA levels in the dorsolateral prefrontal cortex (Emamian et al., 2004; Kozlovsky et al., 2004; Koros and Dorner-Ciossek, 2007).
Psychopharmacological studies provide the most promising evidence in support of the involvement of GSK-3β signaling in schizophrenia. Several studies have reported that chronic administration of antipsychotics phosphorylates GSK-3β at the Ser-9 residue, and inhibits its activity (Emamian et al., 2004; Koros and Dorner-Ciossek, 2007). More specifically, we saw chronic treatment of mice with haloperidol can increase phosphorylation of GSK-3β at Ser-9, which is associated with increased phosphorylation levels of AKT on both Thr-308 and Ser-473 phosphorylation sites in the frontal cortex (Emamian et al., 2004). Other studies also reported that chronic administration of clozapine, risperidone or haloperidol can enhance protein levels of β-catenin and GSK-3 as well as the inhibitory phosphorylation of GSK-3β in the rat prefrontal cortex and striatum (Alimohamad et al., 2005; Kozlovsky et al., 2006). Atypical antipsychotics after acute administration can increase phosphorylation of GSK-3β in the mouse brain (Li et al., 2007). Besides antipsychotics, lithium, and electroconvulsive shocks used to augment antipsychotic treatment in schizophrenic patients have been shown to lead to activation of AKT and concomitant inhibition of GSK-3β in rats (Kang et al., 2004; Gould, 2006).
Modulation of the activity of receptors of several neurotransmitters involved in schizophrenia can affect GSK-3β activity (Koros and Dorner-Ciossek, 2007). Administration of NMDA ligand can dephosphorylate GSK-3β at Ser-9 which leads to its activation (Luo et al., 2003). Consistent with this finding, non-competitive antagonists of NMDA receptors can inhibit GSK-3β by phosphorylation of Ser-9 site (Gould and Manji, 2005). Dopamine receptor antagonists as well as chemicals enhancing serotonergic transmission have been found to increase the expression and inhibitory phosphorylation of GSK-3β and activation of AKT in the mouse brain (Li et al., 2004, 2007).
A Brief Review of other Signaling Pathways Involved in Schizophrenia
Besides the AKT/GSK3 signaling pathway described above, several other signaling molecules have been identified during the last decade to play an important role in schizophrenia. Signaling molecules generally act through a network of proteins to convey a signal from cell membrane to downstream molecules, such as transcription factors in the nucleus. Therefore, signaling molecules are functionally connected to each other and disruption in the function of one molecule could interfere with the efficacy and efficiency of signal transduction from cell membrane to the nucleus, which ultimately can result in the failure of cellular function. In this section DISC1, NRG1, and calcineurin are described as examples of intracellular molecules that are directly associated with schizophrenia through both genetic and functional studies. However, several other known signaling molecules are reported to be involved in schizophrenia but they are not discussed in this review due to the limitation of space.
Disrupted in Schizophrenia-1 (DISC1)
DISC1 is now widely accepted as one of the most important candidate genes for schizophrenia. A wide range of genetic, proteomic, and biochemical studies, have provided strong evidence in support of the involvement of this gene in the development of schizophrenia. This gene was initially reported in a pedigree of a large Scottish family with many individuals displaying a variety of psychiatric diagnoses including schizophrenia, bipolar disorder, recurrent depression, and conduct disorder (St Clair et al., 1990). Genomic analysis of the pedigree revealed the presence of a t(1;11)(q42;q14) balanced chromosomal translocation which cosegregated with mental illness in affected individuals, and lead to a disruption of two genes, Disrupted In Schizophrenia-1 (DISC1) and Disrupted In Schizophrenia-2 (DISC2) (Millar et al., 2000; Kvajo et al., 2010). Linkage and association studies linked DISC1 variants to schizophrenia, schizoaffective disorder, and major depression (Allen et al., 2008). There are four splice variants of the DISC1 gene including the L (Long; 90 kDa), Lv (Long Variant; 95 kDa), S (Short; 70 kDa), and Es (Extremely Short; 41 kDa) (Taylor et al., 2003). The Scottish translocation occurs between exons 8 and 9 of the gene, truncating the C-terminal region of the major (L, Lv, and S) isoforms (Millar et al., 2000). The extremely short DISC1 isoform is present in the Scottish translocation carriers (Taylor et al., 2003; Kvajo et al., 2010).
At the protein level, DISC1 has an N-terminal domain with no clear structure, and a long helical C-terminal domain containing multiple regions with high coiled-coil forming potential, which can bind multiple proteins involved in different cellular processes (Millar et al., 2000). Recent studies provide evidence for the connection between DISC1 and AKT signaling pathways. For example, it has been recently shown that DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212 (Kim et al., 2009). More importantly, it has been shown that DISC1 binds phosphodiesterase 4b (PDE4b), an enzyme which inactivates cAMP, and is believed to be the only way of cAMP degradation in the brain (Millar et al., 2005).
In terms of subcellular localization, DISC1 has been reported to localize in different compartments including the centrosome (Morris et al., 2003), mitochondria (James et al., 2004), spines and dendrites (Millar et al., 2005; Bradshaw et al., 2008), growth cones and axon terminals in neurons and neuronal like cells (Taya et al., 2007), stress granules (Ogawa et al., 2005) and the nucleus (Sawamura et al., 2005). This pattern of localizations lead to a suggestion that DISC1 may act as a molecular scaffold and serve as an assembly point for large protein complexes (Kvajo et al., 2010). During the development of the brain, DISC1 mRNA can be detected in all hippocampal subfields, the cortex, and the thalamus. In the adult brain, however, DISC1 displays a more restricted localization with a high expression only in the dentate gyrus of the hippocampus, and, to a lesser degree in the CA1 region of hippocampus (Austin et al., 2004).
A number of studies in human showed that healthy subjects and patients with schizophrenia carrying DISC1 risk alleles display changes in gray matter volume in the hippocampus, prefrontal cortex, and other brain regions (Callicott et al., 2005; Cannon et al., 2005; Di Giorgio et al., 2008; Szeszko et al., 2008). DISC1 risk alleles are also associated with deficits in working memory, short-term memory, and long-term memory (Burdick et al., 2005; Cannon et al., 2005; Hennah et al., 2005), attention (Hennah et al., 2005; Liu et al., 2006), and hippocampal and prefrontal cortical activation (Callicott et al., 2005; Prata et al., 2008).
Deficiency in DISC1 expression in mice caused region-specific phenotypes, including altered organization and complexity of newly born and mature neurons of the dentate gyrus, as well as changes in short-term plasticity in CA3/CA1 synapses (Kvajo et al., 2008). DISC1 deficient mouse shows highly selective impairment in working memory as well (Koike et al., 2006; Kvajo et al., 2008). Other mouse models of DISC1 have been generated that show selective cognitive behavioral phenotypes (Clapcote et al., 2007; Hikida et al., 2007; Pletnikov et al., 2007, 2008). Overexpression of truncated versions of the human DISC1 also provided evidence for anatomical and behavioral abnormalities (Hikida et al., 2007; Pletnikov et al., 2007, 2008). Moreover, mice carrying point mutations in the DISC1 gene demonstrated that allelic heterogeneity at the DISC1 locus can lead to distinct behavioral phenotypes (Clapcote et al., 2007; Kvajo et al., 2010).
Neuregulin 1 (NRG1)
Several review articles have been already published that discuss the genetic, neurobiological, and pathophysiologic studies of NRG1 and ErbB4 in schizophrenia (Mei and Xiong, 2008; Talmage, 2008; Jaaro-Peled et al., 2009; Banerjee et al., 2010). NRG1 is also one of the most important schizophrenia susceptibility genes, for which risk genotypes or haplotypes have been identified and its associations with the illness have been replicated in different populations (Mei and Xiong, 2008; Banerjee et al., 2010).
NRG1 has diverse neurobiological functions, some of which have been implicated in the pathophysiology of schizophrenia (Mei and Xiong, 2008; Talmage, 2008; Jaaro-Peled et al., 2009; Banerjee et al., 2010). These functions include neuronal migration, neuro-glial trophic effects, myelination and modulation of glutamatergic or GABAergic neurotransmitter systems (Mei and Xiong, 2008; Talmage, 2008; Jaaro-Peled et al., 2009; Banerjee et al., 2010). Moreover, there is genetic evidence for ErbB4, tyrosine kinase receptor, as a candidate susceptibility gene as well as a positive epistatic interactions between NRG1 and ErbB4 in schizophrenia. ErbB4 interacts with NRG1 and influences glutamatergic and GABAergic and neurotrophic transmissions. These neurotransmitter systems have all been implicated in the pathogenesis of schizophrenia. Therefore, there is a significant body of literature supporting the role of NRG1–ErbB4 signaling pathway in the development of schizophrenia (Mei and Xiong, 2008; Talmage, 2008; Jaaro-Peled et al., 2009; Banerjee et al., 2010).
While genetic, neurobiological and endophenotypic investigations support the role of the NRG1–ErbB4 pathway in schizophrenia, there are recent evidence showing that this pathway could act along with other signaling molecules involved in schizophrenia (Banerjee et al., 2010). Positive association of NRG1 with schizophrenia has been replicated in various populations. Some genotype–phenotype association studies have demonstrated links between risk genotypes of NRG1 and some of the disease manifestations of schizophrenia (Banerjee et al., 2010). However, most disease endophenotypes were not associated with the NRG1 or ErbB4 risk haplotypes, but rather with a single nucleotide polymorphism within the hapblocks (Mei and Xiong, 2008; Talmage, 2008; Jaaro-Peled et al., 2009; Banerjee et al., 2010). Interestingly, recent studies by Nicodemus et al. (2010) show evidence of increased schizophrenia risk with NRG1, ErbB4, and AKT1 epistasis via functional neuroimaging in healthy controls (Nicodemus et al., 2010). The same group also reported epistatic and functional interactions of catechol-O-methyltransferase (COMT) and AKT1 on NRG1-ErbB signaling (Sei et al., 2010).
Calcineurin (Protein Phosphatase 2B or PP2B)
Calcineurin is a calcium/calmodulin dependent Ser/Thr phosphatase and is widely expressed in the brain and other tissues. It dephosphorylates a large number of signaling proteins and, therefore, is involved in the regulation of several neuronal processes (Shibasaki et al., 2002). Calcineurin was first linked to schizophrenia by the study of Gerber et al. (2003). Gerber et al. studied a subset of calcineurin-related genes that map to loci previously implicated in schizophrenia by linkage studies and detected association of the PPP3CC gene, which encodes the calcineurin gamma catalytic subunit, with schizophrenia. Some studies could reproduce this finding (Goto et al., 1986; Horiuchi et al., 2007; Liu et al., 2007; Yamada et al., 2007) and some failed to show an association, possibly due to the power of the studies and the level of risk associated with altered calcineurin signaling (Kinoshita et al., 2005; Xi et al., 2007; Sanders et al., 2008; Kvajo et al., 2010). Regardless of genetic association, calcineurin is functionally important because it can affect the activity of several other signaling molecules involved in schizophrenia (described below).
Molecular Mechanisms Underlying the role of AKT/GSK3 Pathway in Schizophrenia
AKT is recruited to the cell membrane by binding to lipids generated by PI3 kinase (Brazil et al., 2004). In response to ligand binding to plasma membrane receptors of different tyrosine kinases, PI3K generates phosphatidylinositol 3,4,5 trisphosphate (PIP3) at the cell surface, which leads to recruitment of AKT to the plasma membrane through its association with the newly generated PIP3 (Brazil et al., 2004). AKT is subsequently activated by phosphorylation at Thr-308, catalyzed by PDK1. A second phosphorylation at Ser-473, by an unknown kinase, can augment its activity up to 10-folds (Brazil et al., 2004). Activated AKT phosphorylates a number of other molecules including the clinically relevant target, GSK3 (Figure 1, Brazil et al., 2004). GSK3 plays several roles in glucose metabolism, differentiation and development, intracellular trafficking, apoptosis, and regulation of gene transcription (Kockeritz et al., 2006). In the brain, some studies suggest that GSK3, like AKT, could modulate synaptic plasticity (Peineau et al., 2008). Unlike AKT that has to be phosphorylated before activation (Brazil et al., 2004), GSK3 is constitutively active in resting cells, requiring phosphorylation by kinases such as AKT to inactivate it (Doble and Woodgett, 2003).
Signaling through Wnt pathway has been also reported to be involved in schizophrenia. The Wnt signaling plays a critical role in a variety of developmental processes during embryonic development but recent studies have also suggested a role for the Wnt pathway in synaptic modulation and plasticity during the adult life (Hall et al., 2000). GSK-3β is an intermediate cytoplasmic component in Wnt pathway (Nelson and Nusse, 2004). GSK-3β forms a complex with multiple proteins such as β-catenin, APC and Axin (Lovestone et al., 2007). GSK-3β constitutively phosphorylates β-catenin, which results in its degradation and the subsequent inhibition of β-catenin-mediated gene transcription. Activation of Wnt receptor facilitates the physical separation of GSK-3β from β-catenin (Lovestone et al., 2007). This separation blocks phosphorylation of β-catenin by GSK-3β, which prevents degradation of β-catenin and causes its nuclear localization. In the nucleus, β-catenin regulates transcription and expression of specific proteins in the cell (Lovestone et al., 2007). Postmortem studies showed decreased in β-catenin levels (Lovestone et al., 2007) and an increased expression of Wnt-1 in the brain samples from patients with schizophrenia (Miyaoka et al., 1999). Taken together, many reports consistently showed the involvement of AKT-GSK3-Wnt pathway in schizophrenia, which makes them an attractive target for development of novel therapeutics for treatment of schizophrenia and other psychiatric disorders (Freyberg et al., 2010).
An interesting study by Mao et al. (2009) reported an interaction between DISC1 and GSK-3β that plays a role in neurogenesis during brain development and adulthood (Mao et al., 2009). Reduction of the expression of DISC1 in neurons decreases neurogenesis mediated by β-catenin signaling (Mao et al., 2009). More specifically, DISC1 can directly bind to GSK-3β and inhibit its activity. This inhibition interferes with its ability to phosphorylate β-catenin. It also affects GSK-3β's auto-phosphorylation at Y216. Interestingly, DISC1-mediated inhibition of GSK-3β affects a selective set of GSK-3β substrates and some of the well-characterized targets apparently are not affected. As discussed above, GSK-3β is immediate downstream molecule to the AKT and a key player of Wnt signaling pathways (Freyberg et al., 2010). This new interaction adds another interesting dimension in support of the convergent evidence for the involvement of AKT/GSK3 signaling pathway in schizophrenia that needs to be dissected out with further studies.
Dopamine receptors have been the center of attention of several groups to investigate how AKT is involved in schizophrenia. Dopamine D2 class receptors (D2−4 receptors) can modulate the activity of AKT (Beaulieu et al., 2007). Activation of dopamine D2 receptor by dopamine leads to recruitment of β-arrestin 2, a scaffolding protein, which facilitates receptor desensitization and the subsequent internalization (Beaulieu et al., 2005). AKT is also recruited into this complex along with the phosphatase PP2A. PP2A dephosphorylates and inactivates AKT, leading to an increased GSK3 activity (Beaulieu et al., 2004, 2005; Klewe et al., 2008; Skinbjerg et al., 2009). Beaulieu et al. also showed that activation of dopamine D2 receptor can inhibit AKT activity through an arrestin-dependent but G protein-independent pathway (Beaulieu et al., 2005, 2007). This interesting observation shed new light on the mechanisms of antipsychotic action of the D2 receptor blockers. In other words, antipsychotic medications may prevent β-arrestin 2 recruitment through blocking dopamine D2 receptor (Klewe et al., 2008; Masri et al., 2008) and the subsequent activation of AKT. The same group has shown similar function for lithium, which is essentially a mood stabilizer not an antipsychotic drug. Lithium has been shown to disrupt the β-arrestin 2/AKT/PP2A complex, thereby preventing dopamine-induced dephosphorylation of AKT, which is associated with reversal of amphetamine-induced locomotion (Beaulieu et al., 2005). Moreover, amphetamine-induced locomotion is greatly diminished in β-arrestin 2 knockout mice, suggesting that this pathway is critical to at least some psychostimulant effects (Beaulieu et al., 2005). Several studies have shown that schizophrenia and bipolar mood disorder (BMD) share several genes and pathways that are critically involved in both disorders. However, the clinical phenotypes of these two disorders are very different. It is not clear how two entirely different classes of psychotropic drugs, i.e., antipsychotics and mood stabilizers, act through similar signaling pathways but produce completely different clinical pictures.
Transcription Factors Related to Schizophrenia
AKT and GSK3 can modulate the activity of several transcription factors; therefore, they can change the expression patterns of many genes that are regulated by those transcription factors. Some transcription factors are reported to be directly associated with schizophrenia. Association between Transcription Factor 4 (TCF4) and schizophrenia has been recently reported, which influences verbal learning and memory functioning in schizophrenia patients (Lennertz et al., 2011; Williams et al., 2011). Variants of the v-rel avian reticuloendotheliosis viral oncogene homolog A (RELA) gene are associated with schizophrenia and their startle responses (Hashimoto et al., 2011). RELA encodes the major component of the nuclear factor kappa B (NF-κ B) complex that has important roles in the immune and inflammatory responses. The association between RELA gene and schizophrenia was evident in male subjects but not in female subjects (Song et al., 2009; Hashimoto et al., 2011). Postmortem studies showed decreased in β-catenin levels (Lovestone et al., 2007) and an increased expression of Wnt-1 in the brain samples from subjects with schizophrenia (Miyaoka et al., 1999). Georgieva et al. (2006) provided convergent evidence for the involvement of transcription factor oligodendrocyte lineage 2 (OLIG2) in schizophrenia. They show variation in OLIG2 confers susceptibility to schizophrenia alone and as part of a network of genes implicated in oligodendrocyte function (Georgieva et al., 2006).
Some important signaling molecules that show association with schizophrenia could affect transcriptional activity of other transcription factors as well. For example, calcineurin (PP2B) can directly regulate the activity of CREB and Nuclear Factor of Activated T cells (NFATc), which are both involved in cognitive functions known to be impaired in schizophrenia. CREB not only is known to be involved in depression (Carlezon et al., 2005), but also some variants in the promoter region of CREB in patients with schizophrenia have been reported as well (Kawanishi et al., 1999). Kawanishi and colleagues analyzed the promoter region of the human CREB in schizophrenics and healthy subjects and found two novel variants (−933T–>C and −413G–>A) only in schizophrenics. Calcineurin can activate the transcription factor NFAT, which has a role in neuronal growth and axonal guidance during development in vitro. Interestingly, NFAT mutant mice shows severe neurodevelopmental defects in vivo (Graef et al., 2001).
Conclusion
Review of the literature published during the past few years provide strong support for the involvement of AKT/GSK3 signaling pathway in the development of schizophrenia. A number of studies strongly suggest targeting this pathway is a promising approach for development of novel psychotropic drugs for treatment of schizophrenia and mood disorders. At the same time, there are several other signaling molecules and pathways that show conclusive evidence for being involved in pathogenesis of schizophrenia. Despite many differences in the approach of scientists working on schizophrenia, they have come to a general consensus that schizophrenia is most likely caused by the altered function or expression of many genes. Such genes may individually contribute only to a small risk, but their cumulative effects cause the dysfunction of brain, which manifests itself by the clinical picture we call schizophrenia. Although there has been significant progress in identification of the role of several signaling molecules in schizophrenia, we still do not know how much each gene contributes to the development of pathology. Therefore, we need to develop novel systems biology tools that can quantify the role of individual or multiple genes in disease development. The recently developed fault diagnosis engineering technology in molecular networks is a promising tool that has such capabilities and can model complex trait disorders such as schizophrenia (Abdi et al., 2008, 2009; Emamian and Abdi, 2009; Abdi and Emamian, 2010).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdi, A., and Emamian, E. S. (2010). Fault diagnosis engineering in molecular signaling networks: an overview and applications in target discovery. Chem. Biodivers. 7, 1111–1123.
Abdi, A., Tahoori, M. B., and Emamian, E. S. (2008). Fault diagnosis engineering of digital circuits can identify vulnerable molecules in complex cellular pathways. Sci. Signal 42, 48–61.
Abdi, A., Tahoori, M. B., and Emamian, E. S. (2009). Identification of critical molecules via fault diagnosis engineering. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2009, 4898–4901.
Akama, K. T., and McEwen, B. S. (2003). Estrogen stimulates postsynaptic density-95 rapid protein synthesis via the AKT/protein kinase B pathway. J. Neurosci. 23, 2333–2339.
Alessi, D. R., Caudwell, F. B., Andjelkovic, M., Hemmings, B. A., and Cohen, P. (1996). Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 399, 333–338.
Alimohamad, H., Rajakumar, N., Seah, Y. H., and Rushlow, W. (2005). Antipsychotics alter the protein expression levels of beta-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol. Psychiatry 57, 533–542.
Allen, N. C., Bagade, S., McQueen, M. B., Ioannidis, J. P., Kavvoura, F. K., Khoury, M. J., Tanzi, R. E., and Bertram, L. (2008). Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat. Genet. 40, 827–834.
Altomare, D. A., Guo, K., Cheng, J. Q., Sonoda, G., Walsh, K., and Testa, J. R. (1995). Cloning, chromosomal localization and expression analysis of the mouse Akt2 oncogene. Oncogene 11, 1055–1060.
Austin, C. P., Ky, B., Ma, L., Morris, J. A., and Shughrue, P. J. (2004). Expression of Disrupted-In-Schizophrenia-1, a schizophrenia-associated gene, is prominent in the mouse hippocampus throughout brain development. Neuroscience 124, 3–10.
Bajestan, S. N., Sabouri, A. H., Nakamura, M., Takashima, H., Keikhaee, M. R., Behdani, F., Fayyazi, M. R., Sargolzaee, M. R., Bajestan, M. N., Sabouri, Z., Khayami, E., Haghighi, S., Hashemi, S. B., Eiraku, N., Tufani, H., Najmabadi, H., Arimura, K., Sano, A., and Osame, M. (2006). Association of AKT1 haplotype with the risk of schizophrenia in Iranian population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141, 383–386.
Banerjee, A., Macdonald, M. L., Borgmann-Winter, K. E., and Hahn, C. G. (2010). Neuregulin 1-erbB4 pathway in schizophrenia: from genes to an interactome. Brain Res. Bull. 83, 132–139.
Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An AKT/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273.
Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., and Caron, M. G. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104.
Beaulieu, J. M., Tirotta, E., Sotnikova, T. D., Masri, B., Salahpour, A., Gainetdinov, R. R., Borrelli, E., and Caron, M. G. (2007). Regulation of AKT signaling by D2 and D3 dopamine receptors in vivo. J. Neurosci. 27, 881–885.
Beffert, U., Morfini, G., Bock, H. H., Reyna, H., Brady, S. T., and Herz, J. (2002). Reelin-mediated signaling locally regulates protein kinase B/AKT and glycogen synthase kinase 3beta. J. Biol. Chem. 277, 49958–49964.
Birnbaum, M. J. (2004). On the InterAKTion between hexokinase and the mitochondrion. Dev. Cell 7, 781–782.
Blain, S. W., and Massague, J. (2002). Breast cancer banishes p27 from nucleus. Nat. Med. 8, 1076–1078.
Bradshaw, N. J., Ogawa, F., Antolin-Fontes, B., Chubb, J. E., Carlyle, B. C., Christie, S., Claessens, A., Porteous, D. J., and Millar, J. K. (2008). DISC1, PDE4B, and NDE1 at the centrosome and synapse. Biochem. Biophys. Res. Commun. 377, 1091–1096.
Brazil, D. P., Yang, Z. Z., and Hemmings, B. A. (2004). Advances in protein kinase B signaling: AKTion on multiple fronts. Trends Biochem. Sci. 29, 233–242.
Burdick, K. E., Hodgkinson, C. A., Szeszko, P. R., Lencz, T., Ekholm, J. M., Kane, J. M., Goldman, D., and Malhotra, A. K. (2005). DISC1 and neurocognitive function in schizophrenia. Neuroreport 16, 1399–1402.
Callicott, J. H., Straub, R. E., Pezawas, L., Egan, M. F., Mattay, V. S., Hariri, A. R., Verchinski, B. A., Meyer-Lindenberg, A., Balkissoon, R., Kolachana, B., Goldberg, T. E., and Weinberger, D. R. (2005). Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 102, 8627–8632.
Cannon, T. D., Hennah, W., van Erp, T. G., Thompson, P. M., Lonnqvist, J., Huttunen, M., Gasperoni, T., Tuulio-Henriksson, A., Pirkola, T., Toga, A. W., Kaprio, J., Mazziotta, J., and Peltonen, L. (2005). Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch. Gen. Psychiatry 62, 1205–1213.
Carlezon, W. A. Jr., Duman, R. S., and Nestler, E. J. (2005). The many faces of CREB. Trends Neurosci. 28, 436–445.
Chen, H. K., Fernandez-Funez, P., Acevedo, S. F., Lam, Y. C., Kaytor, M. D., Fernandez, M. H., Aitken, A., Skoulakis, E. M., Orr, H. T., Botas, J., and Zoghbi, H. Y. (2003). Interaction of AKT-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 113, 457–468.
Chong, Z. Z., Li, F., and Maiese, K. (2005). Activating Akt and the brain's resources to drive cellular survival and prevent inflammatory injury. Histol. Histopathol. 20, 299–315.
Clapcote, S. J., Lipina, T. V., Millar, J. K., Mackie, S., Christie, S., Ogawa, F., Lerch, J. P., Trimble, K., Uchiyama, M., Sakuraba, Y., Kaneda, H., Shiroishi, T., Houslay, M. D., Henkelman, R. M., Sled, J. G., Gondo, Y., Porteous, D. J., and Roder, J. C. (2007). Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54, 387–402.
Colin, E., Regulier, E., Perrin, V., Durr, A., Brice, A., Aebischer, P., Deglon, N., Humbert, S., and Saudou, F. (2005). AKT is altered in an animal model of Huntington's disease and in patients. Eur. J. Neurosci. 21, 1478–1488.
Conery, A. R., Cao, Y., Thompson, E. A., Townsend, C. M. Jr., Ko, T. C., and Luo, K. (2004). Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat. Cell Biol. 6, 366–372.
Costa-Mattioli, M., Sossin, W. S., Klann, E., and Sonenberg, N. (2009). Translational control of long-lasting synaptic plasticity and memory. Neuron 15, 10–26.
Di Giorgio, A., Blasi, G., Sambataro, F., Rampino, A., Papazacharias, A., Gambi, F., Romano, R., Caforio, G., Rizzo, M., Latorre, V., Popolizio, T., Kolachana, B., Callicott, J. H., Nardini, M., Weinberger, D. R., and Bertolino, A. (2008). Association of the SerCys DISC1 polymorphism with human hippocampal formation gray matter and function during memory encoding. Eur. J. Neurosci. 28, 2129–2136.
Doble, B. W., and Woodgett, J. R. (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186.
Dudek, H., Datta, S. R., Franke, T. F., Birnbaum, M. J., Yao, R., Cooper, G. M., Segal, R. A., Kaplan, D. R., and Greenberg, M. E. (1997). Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275, 661–665.
Dummler, B., and Hemmings, B. A. (2007). Physiological roles of PKB/AKT isoforms in development and disease. Biochem. Soc. Trans. 35, 231–235.
Emamian, E. S., and Abdi, A. (2009). Complex human disorders and molecular system engineering: historical perspective and potential impacts. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2009, 1083–1085.
Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M., and Gogos, J. A. (2004). Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat. Genet. 36, 131–137.
Emamian, E. S., Kaytor, M. D., Duvick, L. A., Zu, T., Tousey, S. K., Zoghbi, H. Y., Clark, H. B., and Orr, H. T. (2003). Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38, 375–387.
Freyberg, Z., Ferrando, S. J., and Javitch, J. A. (2010). Roles of the AKT/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am. J. Psychiatry 167, 388–396.
Georgieva, L., Moskvina, V., Peirce, T., Norton, N., Bray, N. J., Jones, L., Holmans, P., Macgregor, S., Zammit, S., Wilkinson, J., Williams, H., Nikolov, I., Williams, N., Ivanov, D., Davis, K. L., Haroutunian, V., Buxbaum, J. D., Craddock, N., Kirov, G., Owen, M. J., and O'Donovan, M. C. (2006). Convergent evidence that oligodendrocyte lineage transcription factor 2 (OLIG2) and interacting genes influence susceptibility to schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 103, 12469–12474.
Gerber, D. J., Hall, D., Miyakawa, T., Demars, S., Gogos, J. A., Karayiorgou, M., and Tonegawa, S. (2003). Evidence for association of schizophrenia with genetic variation in the 8p21.3 gene, PPP3CC, encoding the calcineurin gamma subunit. Proc. Natl. Acad. Sci. U.S.A. 100, 8993–8998.
Gonzalez, E., and McGraw, T. E. (2009). Insulin-modulated AKT subcellular localization determines AKT isoform-specific signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 7004–7009.
Goto, S., Matsukado, Y., Mihara, Y., Inoue, N., and Miyamoto, E. (1986). The distribution of calcineurin in rat brain by light and electron microscopic immunohistochemistry and enzyme-immunoassay. Brain Res. 397, 161–172.
Graef, I. A., Chen, F., Chen, L., Kuo, A., and Crabtree, G. R. (2001). Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 105, 863–875.
Greengard, P., Valtorta, F., Czernik, A. J., and Benfenati, F. (1993). Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 259, 780–785.
Gould, T. D. (2006). Targeting glycogen synthase kinase-3 as an approach to develop novel mood-stabilising medications. Expert. Opin. Ther. Targets 10, 377–392.
Gould, T. D., and Manji, H. K. (2005). Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology 30, 1223–1237.
Griffin, R. J., Moloney, A., Kelliher, M., Johnston, J. A., Ravid, R., Dockery, P., O'Connor, R., and O'Neill, C. (2005). Activation of AKT/PKB, increased phosphorylation of AKT substrates and loss and altered distribution of AKT and PTEN are features of Alzheimer's disease pathology. J. Neurochem. 93, 105–117.
Guo, H. F., and Zhong, Y. (2006). Requirement of AKT to mediate long-term synaptic depression in Drosophila. J. Neurosci. 12, 4004–4014.
Hall, A. C., Lucas, F. R., and Salinas, P. C. (2000). Axonal remodelling and synaptic differentiation in the cerebellum is regulated by Wnt-7a signaling. Cell 100, 525–535.
Hashimoto, R., Ohi, K., Yasuda, Y., Fukumoto, M., Yamamori, H., Takahashi, H., Iwase, M., Okochi, T., Kazui, H., Saitoh, O., Tatsumi, M., Iwata, N., Ozaki, N., Kamijima, K., Kunugi, H., and Takeda, M. (2011). Variants of the RELA gene are associated with schizophrenia and their startle responses. Neuropsychopharmacology 36, 1921–1931.
Hennah, W., Tuulio-Henriksson, A., Paunio, T., Ekelund, J., Varilo, T., Partonen, T., Cannon, T. D., Lonnqvist, J., and Peltonen, L. (2005). A haplotype within the DISC1 gene is associated with visual memory functions in families with a high density of schizophrenia. Mol. Psychiatry 10, 1097–1103.
Henry, M. K., Lynch, J. T., Eapen, A. K., and Quelle, F. W. (2001). DNA damage-induced cell-cycle arrest of hematopoietic cells is overridden by activation of the PI-3 kinase/Akt signaling pathway. Blood 98, 834–841.
Hikida, T., Jaaro-Peled, H., Seshadri, S., Oishi, K., Hookway, C., Kong, S., Wu, D., Xue, R., Andrade, M., Tankou, S., Mori, S., Gallagher, M., Ishizuka, K., Pletnikov, M., Kida, S., and Sawa, A. (2007). From the cover: dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc. Natl. Acad. Sci. U.S.A. 104, 14501–14506.
Horiuchi, Y., Ishiguro, H., Koga, M., Inada, T., Iwata, N., Ozaki, N., Ujike, H., Muratake, T., Someya, T., and Arinami, T. (2007). Support for association of the PPP3CC gene with schizophrenia. Mol. Psychiatry 12, 891–893.
Horváth, S., Janka, Z., and Mirnics, K. (2011). Analyzing schizophrenia by DNA microarrays. Biol. Psychiatry 15, 157–162.
Hou, L., and Klann, E. (2004). Activation of the phosphoinositide 3-kinase-AKT-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 4, 6352–6361.
Ikeda, M., Iwata, N., Suzuki, T., Kitajima, T., Yamanouchi, Y., Kinoshita, Y., Inada, T., and Ozaki, N. (2004). Association of AKT1 with schizophrenia confirmed in a Japanese population. Biol. Psychiatry 56, 698–700.
Jaaro-Peled, H., Hayashi-Takagi, A., Seshadri, S., Kamiya, A., Brandon, N. J., and Sawa, A. (2009). Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 32, 485–495.
James, R., Adams, R. R., Christie, S., Buchanan, S. R., Porteous, D. J., and Millar, J. K. (2004). Disrupted in Schizophrenia 1 (DISC1) is a multicompartmentalized protein that predominantly localizes to mitochondria. Mol. Cell. Neurosci. 26, 112–122.
Jonassen, A. K., Sack, M. N., Mjos, O. D., and Yellon, D. M. (2001). Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ. Res. 89, 1191–1198.
Kandel, E. S., and Hay, N. (1999). The regulation and activities of the multifunctional serine/threonine kinase AKT/PKB. Exp. Cell Res. 253, 210–229.
Kang, J. Q., Chong, Z. Z., and Maiese, K. (2003a). Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J. Neurosci. Res. 74, 37–51.
Kang, J. Q., Chong, Z. Z., and Maiese, K. (2003b). Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol. Pharmacol. 64, 557–569.
Kang, U. G., Roh, M. S., and Jung, J. R. (2004). Activation of protein kinase B (Akt) signaling after electroconvulsive shock in the rat hippocamous. Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 41–44.
Kaspar, B. K., Llado, J., Sherkat, N., Rothstein, J. D., and Gage, F. H. (2003). Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301, 839–842.
Kawanishi, Y., Harada, S., Tachikawa, H., Okubo, T., and Shiraishi, H. (1999). Novel variants in the promoter region of the CREB gene in schizophrenic patients. J. Hum. Genet. 44, 428–430.
Kim, J. Y., Duan, X., Liu, C. Y., Jang, M. H., Guo, J. U., Pow-anpongkul, N., Kang, E., Song, H., and Ming, G. L. (2009). DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 63, 761–773.
Kinoshita, Y., Suzuki, T., Ikeda, M., Kitajima, T., Yamanouchi, Y., Inada, T., Yoneda, H., Iwata, N., and Ozaki, N. (2005). No association with the calcineurin A gamma subunit gene (PPP3CC) haplotype to Japanese schizophrenia. J. Neural Transm. 112, 1255–1262.
Klann, E., and Dever, T. E. (2004). Biochemical mechanisms for translational regulation in synaptic plasticity. Nat. Rev. Neurosci. 5, 931–942.
Klewe, I. V., Nielsen, S. M., Tarpo, L., Urizar, E., Dipace, C., Javitch, J. A., Gether, U., Egebjerg, J., and Christensen, K. V. (2008). Recruitment of β-arrestin2 to the dopamine D2 receptor: insights into anti-psychotic and anti-parkinsonian drug receptor signaling. Neuropharmacology 54, 1215–1222.
Kockeritz, L., Doble, B., Patel, S., and Woodgett, J. R. (2006). Glycogen synthase kinase-3-an overview of an over-achieving protein kinase. Curr. Drug Targets 7, 1377–1138.
Koike, H., Arguello, P. A., Kvajo, M., Karayiorgou, M., and Gogos, J. A. (2006). Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc. Natl. Acad. Sci. U.S.A. 103, 3693–3697.
Koros, E., and Dorner-Ciossek, C. (2007). The role of glycogen synthase kinase-3beta in schizophrenia. Drug News Perspect. 20, 437–445.
Kozlovsky, N., Amar, S., Belmaker, R. H., and Agam, G. (2006). Psychotropic drugs affect Ser9-phosphorylated GSK-3 beta protein levels in rodent frontal cortex. Int. J. Neuropsychopharmacol. 9, 337–342.
Kozlovsky, N., Shanon-Weickert, C., Tomaskovic-Crook, E., Kleinman, J. E., Belmaker, R. H., and Agam, G. (2004). Reduced GSK-3beta mRNA levels in post-mortem dorsolateral prefrontal cortex of schizophrenic patients. J. Neural Transm. 111, 1583–1592.
Kvajo, M., McKellar, H., Arguello, P. A., Drew, L. J., Moore, H., Macdermott, A. B., Karayiorgou, M., and Gogos, J. A. (2008). A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc. Natl. Acad. Sci. U.S.A. 105, 7076–7081.
Kvajo, M., McKellar, H., and Gogos, J. A. (2010). Molecules, signaling, and schizophrenia. Curr. Top. Behav. Neurosci. 4, 629–656.
Lai, W. S., Xu, B., Westphal, K. G. C., Paterlini, M., Olivier, B., Pavlidis, P., Karayiorgou, M., and Gogos, J. (2006). AKT1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proc. Natl. Acad. Sci. U.S.A. 103, 16906–16911.
Lee, C. C., Huang, C. C., Wu, M. Y., and Hsu, K. S. (2005). Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J. Biol. Chem. 208, 18543–18550.
Lennertz, L., Rujescu, D., Wagner, M., Frommann, I., Schulze-Rauschenbach, S., Schuhmacher, A., Landsberg, M. W., Franke, P., Möller, H. J., Wölwer, W., Gaebel, W., Häfner, H., Maier, W., and Mössner, R. (2011). Novel schizophrenia risk gene TCF4 influences verbal learning and memory functioning in schizophrenia patients. Neuropsychobiology 63, 131–136.
Li, X., Rosborough, K. M., Friedman, A. B., Zhu, W., and Roth, K. A. (2007). Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int. J. Neuropsychopharmacol. 10, 7–19.
Li, X., Zhu, W., Roh, M. S., Friedman, A. B., Rosborough, K., and Jope, R. S. (2004). In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacology 29, 1426–1431.
Liang, J., Zubovitz, J., Petrocelli, T., Kotchetkov, R., Connor, M. K., Han, K., Lee, J. H., Ciarallo, S., Catzavelos, C., Beniston, R., Franssen, E., and Slingerland, J. M. (2002). PKB/AKT phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 8, 1153–1160.
Lin, C. H., Yeh, S. H., Lin, C. H., Lu, K. T., Leu, T. H., Chang, W. C., and Gean, P. W. (2001). A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron 31, 841–851.
Liu, Y. L., Fann, C. S., Liu, C. M., Chang, C. C., Yang, W. C., Hung, S. I., Yu, S. L., Hwang, T. J., Hsieh, M. H., Liu, C. C., Tsuang, M. M., Wu, J. Y., Jou, Y. S., Faraone, S. V., Tsuang, M. T., Chen, W. J., and Hwu, H. G. (2007). More evidence supports the association of PPP3CC with schizophrenia. Mol. Psychiatry 12, 966–974.
Liu, Y. L., Fann, C. S., Liu, C. M., Chen, W. J., Wu, J. Y., Hung, S. I., Chen, C. H., Jou, Y. S., Liu, S. K., Hwang, T. J., Hsieh, M. H., Ouyang, W. C., Chan, H. Y., Chen, J. J., Yang, W. C., Lin, C. Y., Lee, S. F., and Hwu, H. G. (2006). A single nucleotide polymorphism fine mapping study of chromosome 1q42.1 reveals the vulnerability genes for schizophrenia, GNPAT and DISC1: association with impairment of sustained attention. Biol. Psychiatry 60, 554–562.
Lovestone, S., Killick, R., Forti, M. D., and Murray, R. (2007). Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 30, 142–149.
Luo, H. R., Hattori, H., Hossain, M. A., Hester, L., Huang, Y., Lee-Kwon, W., Donowitz, M., Nagata, E., and Snyder, S. H. (2003). Akt as a mediator of cell death. Proc. Natl. Acad. Sci. U.S.A. 100, 11712–11717.
Maiese, K., and Chong, Z. Z. (2004). Insights into oxidative stress and potential novel therapeutic targets for Alzheimer disease. Restor. Neurol. Neurosci. 22, 87–104.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., Tassa, C., Berry, E. M., Soda, T., Singh, K. K., Biechele, T., Petryshen, T. L., Moon, R. T., Haggarty, S. J., and Tsai, L. H. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031.
Markus, A., Zhong, J., and Snider, W. D. (2002). Raf and akt mediate distinct aspects of sensory axon growth. Neuron 35, 65–76.
Masri, B., Salahpour, A., Didrikson, M., Ghisi, V., Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2008). Antagonism of dopamine D2 receptors/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. U.S.A. 105, 13656–13661.
Matsuzaki, H., Tamatani, M., Mitsuda, N., Namikawa, K., Kiyama, H., Miyake, S., and Tohyama, M. (1999). Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J. Neurochem. 73, 2037–2046.
Mei, L., and Xiong, W. C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9, 437–452.
Millar, J. K., Pickard, B. S., Mackie, S., James, R., Christie, S., Buchanan, S. R., Malloy, M. P., Chubb, J. E., Huston, E., Baillie, G. S., Thomson, P. A., Hill, E. V., Brandon, N. J., Rain, J. C., Camargo, L. M., Whiting, P. J., Houslay, M. D., Blackwood, D. H., Muir, W. J., and Porteous, D. J. (2005). DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 310, 1187–1191.
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., Devon, R. S., Clair, D. M., Muir, W. J., Blackwood, D. H., and Porteous, D. J. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423.
Miyaoka, T., Seno, H., and Ishino, H. (1999). Increased expression of Wnt-1 in schizophrenic brains. Schizophr. Res. 38, 1–6.
Mizuno, M., Yamada, K., Takei, N., Tran, M. H., He, J., Nakajima, A., Nawa, H., and Nabeshima, T. (2003). Phosphatidylinositol 3-kinase: a molecule mediating BDNF-dependent spatial memory formation. Mol. Psychiatry 8, 217–224.
Morris, J. A., Kandpal, G., Ma, L., and Austin, C. P. (2003). DISC1 (Disrupted-In-Schizophrenia 1) is a centrosome-associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: regulation and loss of interaction with mutation. Hum. Mol. Genet. 12, 1591–1608.
Namikawa, K., Honma, M., Abe, K., Takeda, M., Mansur, K., Obata, T., Miwa, A., Okado, H., and Kiyama, H. (2000). AKT/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. J. Neurosci. 20, 2875–2886.
Nelson, W. J., and Nusse, R. (2004). Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303, 1483–1487.
Nicodemus, K. K., Law, A. J., Radulescu, E., Luna, A., Kolachana, B., Vakkalanka, R., Rujescu, D., Giegling, I., Straub, R. E., McGee, K., Gold, B., Dean, M., Muglia, P., Callicott, J. H., Tan, H. Y., and Weinberger, D. R. (2010). Biological validation of increased schizophrenia risk with NRG1, ERBB4, and AKT1 epistasis via functional neuroimaging in healthy controls. Arch. Gen. Psychiatry 67, 991–1001.
Niizuma, K., Endo, H., and Chan, P. H. (2009). Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J. Neurochem. 109, 133–138.
Ogawa, F., Kasai, M., and Akiyama, T. (2005). A functional link between Disrupted-In-Schizophrenia 1 and the eukaryotic translation initiation factor 3. Biochem. Biophys. Res. Commun. 338, 771–776.
Ohtsuki, T., Inada, T., and Arinami, T. (2004). Failure to confirm association between AKT1 haplotype and schizophrenia in a Japanese case-control population. Mol. Psychiatry 9, 981–983.
Owada, Y., Utsunomiya, A., Yoshimoto, T., and Kondo, H. (1997). Expression of mRNA for Akt, serine-threonine protein kinase, in the brain during development and its transient enhancement following axotomy of hypoglossal nerve. J. Mol. Neurosci. 9, 27–33.
Papapetropoulos, A., Fulton, D., Mahboubi, K., Kalb, R. G., O'Connor, D. S., Li, F., Altieri, D. C., and Sessa, W. C. (2000). Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/surviving pathway. J. Biol. Chem. 275, 9102–9105.
Peineau, S., Bradley, C., Taghibiglou, C., Doherty, A., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2008). The role of GSK-3 in synaptic plasticity. Br. J. Psychopharmacol. 153, S428–S437.
Pletnikov, M. V., Ayhan, Y., Nikolskaia, O., Xu, Y., Ovanesov, M. V., Huang, H., Mori, S., Moran, T. H., and Ross, C. A. (2007). Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol. Psychiatry 13, 173–186.
Pletnikov, M. V., Ayhan, Y., Xu, Y., Nikolskaia, O., Ovanesov, M., Huang, H., Mori, S., Moran, T. H., and Ross, C. A. (2008). Enlargement of the lateral ventricles in mutant DISC1 transgenic mice. Mol. Psychiatry 13, 115–115.
Prata, D. P., Mechelli, A., Fu, C. H., Picchioni, M., Kane, F., Kalidindi, S., McDonald, C., Kravariti, E., Toulopoulou, T., Miorelli, A., Murray, R., Collier, D. A., and McGuire, P. K. (2008). Effect of disrupted-in schizophrenia-1 on pre-frontal cortical function. Mol. Psychiatry 13, 915–917.
Rytomaa, M., Lehmann, K., and Downward, J. (2000). Matrix detachment induces caspase-dependent cytochrome c release from mitochondria: inhibition by PKB/Akt but not Raf signalling. Oncogene 19, 4461–4468.
Sadock, B. J., Sadock, V. A., Ruiz, P., and Kaplan, H. I. (2009). Kaplan and Sadock's Comprehensive Textbook of Psychiatry. Philadelphia, PA: Ovid Technologies, Inc. Williams and Wilkins.
Sanders, A. R., Duan, J., Levinson, D. F., Shi, J., He, D., Hou, C., Burrell, G. J., Rice, J. P., Nertney, D. A., Olincy, A., Rozic, P., Vinogradov, S., Buccola, N. G., Mowry, B. J., Freedman, R., Amin, F., Black, D. W., Silverman, J. M., Byerley, W. F., Crowe, R. R., Cloninger, C. R., Martinez, M., and Gejman, P. V. (2008). No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: implications for psychiatric genetics. Am. J. Psychiatry 165, 497–506.
Saudou, F., Finkbeiner, S., Devys, D., and Greenberg, M. E. (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66.
Sawamura, N., Sawamura-Yamamoto, T., Ozeki, Y., Ross, C. A., and Sawa, A. (2005). A form of DISC1 enriched in nucleus: altered subcellular distribution in orbitofrontal cortex in psychosis and substance/alcohol abuse. Proc. Natl. Acad. Sci. U.S.A. 102, 118711–118792.
Schwab, S. G., Hoefgen, B., Hanses, C., Hassenbach, M. B., Albus, M., Lerer, B., Trixler, M., Maier, W., and Wildenauer, D. B. (2005). Further evidence for association of variants in the AKT1 gene with schizophrenia in a sample of European sib-pair families. Biol. Psychiatry 58, 446–450.
Sei, Y., Li, Z., Song, J., Ren-Patterson, R., Tunbridge, E. M., Iizuka, Y., Inoue, M., Alfonso, B. T., Beltaifa, S., Nakai, Y., Kolachana, B. S., Chen, J., and Weinberger, D. R. (2010). Epistatic and functional interactions of catechol-o-methyltransferase (COMT) and AKT1 on neuregulin1-ErbB signaling in cell models. PLoS One. 5:e10789. doi: 10.1371/journal.pone.0010789
Shibasaki, F., Hallin, U., and Uchino, H. (2002). Calcineurin as a multifunctional regulator. J. Biochem. 131, 1–15.
Shin, I., Yakes, F. M., Rojo, F., Shin, N. Y., Bakin, A. V., Baselga, J., and Arteaga, C. L. (2002). PKB/AKT mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat. Med. 8, 1145–1152.
Skinbjerg, M., Ariano, M. A., Thorsell, A., Heilig, M., Halldin, C., Innis, R. B., and Sibley, D. R. (2009). Arrestin3 mediates D2 dopamine receptor internalization. Synapse 63, 621–624.
Song, X. Q., Lv, L. X., Li, W. Q., Hao, Y. H., and Zhao, J. P. (2009). The interaction of nuclear factor-kappa B and cytokines is associated with schizophrenia. Biol. Psychiatry 65, 481–488.
St Clair, C. D., Blackwood, D., Muir, W., Carothers, A., Walker, M., Spowart, G., Gosden, C., and Evans, H. J. (1990). Association within a family of a balanced autosomal translocation with major mental illness. Lancet 336, 13–16.
Stilo, S. A., and Murray, R. M. (2010). The epidemiology of schizophrenia: replacing dogma with knowledge. Dialogues Clin. Neurosci. 12, 305–315.
Suhara, T., Mano, T., Oliveira, B. E., and Walsh, K. (2001). Phosphatidylinositol 3-kinase/Akt signaling controls endothelial cell sensitivity to Fas-mediated apoptosis via regulation of FLICE-inhibitory protein (FLIP). Circ. Res. 89, 13–19.
Sui, L., Wang, J., and Li, B. M. (2008). Role of the phosphoinositide 3-kinase-AKT-mammalian target of the rapamycin signaling pathway in long-term potentiation and trace fear conditioning memory in rat medial prefrontal cortex. Learn. Mem. 15, 762–776.
Szeszko, P. R., Hodgkinson, C. A., Robinson, D. G., DeRosse, P., Bilder, R. M., Lencz, T., Burdick, K. E., Napolitano, B., Betensky, J. D., Kane, J. M., Goldman, D., and Malhotra, A. K. (2008). DISC1 is associated with prefrontal cortical gray matter and positive symptoms in schizophrenia. Biol. Psychol. 79, 103–110.
Tan, H. Y., Nicodemus, K. K., Chen, Q., Li, Z., Brooke, J. K., Honea, R., Kolachana, B. S., Straub, R. E., Meyer-Lindenberg, A., Sei, Y., Mattay, V. S., Callicott, J. H., and Weinberger, D. R. (2008). Genetic variation in AKT1 is linked to dopamine-associated prefrontal cortical structure and function in humans. J. Clin. Invest. 118, 2200–2208.
Taya, S., Shinoda, T., Tsuboi, D., Asaki, J., Nagai, K., Hikita, T., Kuroda, S., Kuroda, K., Shimizu, M., Hirotsune, S., Iwamatsu, A., and Kaibuchi, K. (2007). DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J. Neurosci. 27, 15–26.
Taylor, M. S., Devon, R. S., Millar, J. K., and Porteous, D. J. (2003). Evolutionary constraints on the disrupted in schizophrenia locus. Genomics 81, 67–77.
Tiwari, A. K., Zai, C. C., Müller, D. J., and Kennedy, J. L. (2010). Genetics in schizophrenia: where are we and what next? Dialogues Clin. Neurosci. 12, 289–303.
Thiselton, D. L., Vladimirov, V. I., Kuo, P. H., McClay, J., Wormley, B., Fanous, A., O'Neill, F. A., Walsh, D., Van den Oord, J. C. G., Kendler, K. S., and Riley, B. P. (2008). AKT1 is associated with schizophrenia across multiple symptom dimensions in the Irish study of high density schizophrenia families. Biol. Psychiatry 63, 449–457.
Tschopp, O., Yang, Z. Z., Brodbeck, D., Dummler, B. A., Hemmings-Mieszczak, M., Watanabe, T., Michaelis, T., Frahm, J., and Hemmings, B. A. (2005). Essential role of protein kinase B{gamma} (PKB{gamma}/AKT3) in postnatal brain development but not in glucose homeostasis. Development 132, 2943–2954.
Viglietto, G., Motti, M. L., Bruni, P., Melillo, R. M., D'Alessio, A., Califano, D., Vinci, F., Chiappetta, G., Tsichlis, P., Bellacosa, A., Fusco, A., and Santoro, M. (2002). Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/AKT-mediated phosphorylation in breast cancer. Nat. Med. 8, 1136–1144.
Wang, Q., Liu, L., Pei, L., Ju, W., Ahmadian, G., Lu, J., Wang, Y., Liu, F., and Wang, Y. T. (2003). Control of synaptic strength, a novel function of Akt. Neuron 38, 915–928.
Williams, H. J., Moskvina, V., Smith, R. L., Dwyer, S., Russo, G., Owen, M. J., and O'Donovan, M. C. (2011). Association between TCF4 and schizophrenia does not exert its effect by common nonsynonymous variation or by influencing cis-acting regulation of mRNA expression in adult human brain. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156, 781–784.
Williams, H. J., Owen, M. J., and O'Donovan, M. C. (2009). Schizophrenia genetics: new insights from new approaches. Br. Med. Bull. 91, 61–74.
Xi, Z., Yu, L., Shi, Y., Zhang, J., Zheng, Y., He, G., He, L., Wei, Q., Yao, W., Zhang, K., Gu, N., Feng, G., and Zhu, S. (2007). No association between PPP3CC and schizophrenia in the Chinese population. Schizophr. Res. 90, 357–359.
Xu, M. Q., Xing, Q. H., Zheng, Y. L., Li, S., Gao, J. J., He, G., Guo, T. W., Feng, G. Y., Xu, F., and He, L. (2007). Association of AKT1 gene polymorphisms with risk of schizophrenia and with response to antipsychotics in the Chinese population. J. Clin. Psychiatry 68, 1358–1367.
Yamada, K., Gerber, D. J., Iwayama, Y., Ohnishi, T., Ohba, H., Toyota, T., Aruga, J., Minabe, Y., Tonegawa, S., and Yoshikawa, T. (2007). Genetic analysis of the calcineurin pathway identifies members of the EGR gene family, specifically EGR3, as potential susceptibility candidates in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 104, 2815–2820.
Yang, S. D., Yu, J. S., Lee, T. T., Yang, C. C., Ni, M. H., and Yang, Y. Y. (1995). Dysfunction of protein kinase FA/GSK-3 alfa in lymphocytes of patients with schizophrenic disorder. J. Cell. Biochem. 59, 108–116.
Keywords: schizophrenia, neuronal signaling, phosphorylation, AKT, PKB, GSK3
Citation: Emamian ES (2012) AKT/GSK3 signaling pathway and schizophrenia. Front. Mol. Neurosci. 5:33. doi: 10.3389/fnmol.2012.00033
Received: 20 September 2011; Accepted: 25 February 2012;
Published online: 15 March 2012.
Edited by:
Jean-Martin Beaulieu, Duke University Medical Center, USAReviewed by:
Claudia Bagni, Catholic University of Leuven Medical School, BelgiumXiaohua Li, University of Alabama at Birmingham, USA
Ali Salahpour, University of Toronto, Canada
Copyright: © 2012 Emamian. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Effat S. Emamian, Advanced Technologies for Novel Therapeutics, 211 Warren Street, Newark, NJ 07103, USA. e-mail: emame@atnt-usa.com