


Abstract
There is mounting evidence indicating that overexpression or aberrant processing of amyloid precursor protein (βAPP) is causally related to Alzheimer’s disease. βAPP is principally cleaved within the amyloid β protein domain to release a large soluble ectodomain (βAPPs) that has been known to have a wide range of trophic and protective functions. Activation of phospholipase C-coupled receptors has been shown to increase the release of βAPPs through protein kinase C and calcium. Here we have examined whether nicotine can modulate the expression and processing of βAPP in PC12 cells. Treatment of PC12 cells with nicotine increased the release of a carboxyl-terminally truncated, secreted form of βAPP into the conditioned medium without affecting the expression level of βAPP mRNA. The effect of nicotine on the secretion of βAPPs is concentration (>50 μm)- and time (>2 hr)-dependent and attenuated by cotreatment with either mecamylamine, a specific nicotinic receptor antagonist, or EGTA, a calcium chelator, indicating calcium entry through the neuronal nicotinic acetylcholine receptor is essential in enhanced βAPPs release by nicotine. However, nicotine did not significantly change the amyloid β protein secretion from Swedish mutant βAPP-transfected PC12 cells. These results imply that nicotinic receptor agonist might be beneficial in the treatment of Alzheimer’s disease by not only supplementing the deficient cholinergic neurotransmission but also stimulating the release of βAPPs.
AD is characterized by excessive deposition of neuritic plaques and neurofibrillary tangles in the brain. Several lines of evidence indicate that Aβ, which is a principal constituent of neuritic plaques, plays an important role in the pathogenesis of AD (1). Aβ is 39–43 amino acids long and proteolytically derived from an integral membrane protein termed βAPP.
βAPP is normally processed by at least two alternative pathways (2). In a constitutive-secretory pathway, an as yet unidentified enzyme dubbed α secretase cleaves βAPP just outside the membrane to secrete the large extracellular portion (βAPPs) that is known to have potent neurotrophic and neuroprotective activities by stabilizing the intracellular free Ca2+ levels (3). Furthermore, because this α secretase cleavage occurs within the Aβ region, it precludes Aβ formation. However, small amounts of Aβ are normally generated by sequential actions of other unidentified proteases, β and γ secretases. Aβ formation is believed to involve the coated pit-mediated reinternalization of βAPP, which is independent of the constitutive pathway. Overexpression as well as aberrant processing of βAPP can accelerate the secretion of Aβ, which self-aggregates and exerts toxic effects on neurons (1).
Various agents have been shown to regulate the processing of βAPP (2). Especially activation of several PLC-coupled receptors, such as muscarinic m1 and m3 (4, 5), bradykinin (6), thrombin (7), metabotropic glutamate (8), and serotonin 5-HT2a and 5-HT2c receptors (9), has been demonstrated to increase the production of βAPPs and sometimes concomitantly lower the production of Aβ (5, 7). Interestingly, activation of either m2 or m4 muscarinic receptors that are not coupled to PLC but are linked to adenylate cyclase failed to exert such effects (4). The effects of PLC-coupled receptors on βAPP processing are thought to be mainly mediated by PKC, which is stimulated by diacylglycerol formed by PLC activation. Various PKC activators (10, 11) have similar effects on βAPP processing. The effects of PKC on βAPP processing are not due to direct phosphorylation of βAPP but probably to increased formation of βAPP-containing secretory vesicles from the trans-Golgi network (12). PLC activation could also lead to inositol 1,4,5-triphosphate production, thereby raising intracellular calcium levels. The rise in intracellular Ca2+ was also shown to increase βAPPs release in a PKC-independent manner (13, 14). However, the modulation of βAPP processing by ionotropic receptor itself such as nicotinic receptor, which is permeable to Ca2+, has not been pursued in detail.
nAChR is a ligand-gated ion channel and consists of at least eight α-like subunit isoforms(α2–α9) and three β-like subunit isoforms(β2–β4) that exhibit distinct temporal and tissue-specific patterns of expression (15). The α2–α6 subunits require the presence of β subunits to form a functional receptor, whereas the α7–α9 subunits are capable of forming functional channels as homo-oligomers when expressed in Xenopus laevis oocytes or in stably transfected cell lines (15). Neuronal nAChR channels characteristically have a greater Ca2+permeability than muscle nAChR and have been found to elicit diverse behavioral effects including arousal, attention, anxiolytic activity, analgesia, and cognitive enhancement (16). Involvement of nicotinic neurotransmission in cognitive functions is further substantiated by observed deficits in cognitive performance after administration of mecamylamine, a nicotinic receptor antagonist, to humans. Moreover, many studies have indicated a substantial loss of nicotinic receptor population in the brains of AD patients (17), and the degree of cognitive impairments in AD has been reported to correlate well with the central cholinergic deficits (18). In addition, there are epidemiological data showing a negative correlation between smoking and the onset of AD (19), and pilot clinical data indicated that acutely administered nicotine might be beneficial for the treatment of the deficits in attention and information processing associated with AD (20, 21).
In this study, we examined the effects of nicotinic receptor activation on the expression and processing of βAPP in PC12 cells. Treatment of PC12 cells with nicotine increased the secretion of carboxyl-terminally truncated βAPPs into the conditioned medium in a concentration- and time-dependent manner without affecting the levels of βAPP mRNA. The effect of nicotine on the secretion of βAPPs is attenuated by cotreatment with mecamylamine, a specific nicotinic receptor antagonist and EGTA, a calcium chelator.
Materials and Methods
PC12 cells, originated from rat pheochromocytoma, were plated in polyethylenimine-coated 100-mm culture dish at a density of 10,000 cells/cm2 and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and 5% horse serum. Five days after plating, the media were changed to serum-free media and various concentrations of nicotine in the presence or absence of mecamylamine or EGTA were added to the cultures. After incubation with the drugs for the indicated periods, conditioned media were collected, and cells were lyzed for subsequent analyses.
Collected media were centrifuged at 12,000 × g for 15 min to remove the cellular debris and dialyzed against ice-cold buffer consisting of 5 mm Tris·Cl, pH 7.5, and 14 mmNaCl after which the samples were condensed in a lyophilizer and resuspended in an extraction buffer (50 mm Tris, 150 mm NaCl, 5 mm EDTA, pH 7.6, 2% Nonidet P-40, 2% Triton X-100). Cells were harvested in lysis buffer (150 mm NaCl, 50 mm Tris-Cl, pH 8.0, 1 mm EDTA, 1% Nonidet P-40, 100 μg/ml phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin) for 10 min on ice with gentle agitation. The cell lysates were centrifuged for 5 min at 10,000 ×g, and the supernatants were saved at −20° until use. The protein amount in each sample was determined by the Bradford method (Bio-Rad, Hercules, CA). Equal amounts of either cell lysates or resuspended secreted proteins were run on 5–14% gradient SDS- PAGE and transferred to a polyvinylidene fluoride membrane (Bio-Rad). Membranes were blocked by incubation with 6% non-fat dry milk in Tris-buffered saline (20 mm Tris·Cl, 137 mmNaCl, pH 7.6) containing 0.15% Tween-20 overnight at 4° and probed with 22C11 (5 μg/ml; against the 66–81 residues of βAPP; Boehringer Mannheim, Mannheim, Germany), C8 (1:500 dilution; against the last 20 residues of βAPP) (22) or CT15 (1:500 dilution; against the last 15 residues of βAPP) (23) antibody. For preadsorption, 22C11 antibody was incubated with 25 μg/ml purified APLP2 for 1 hr at 4° and then added to the blots. After incubation for 1 hr with gentle agitation at room temperature, blots were reacted with the horseradish peroxidase-coupled goat anti-mouse or anti-rabbit immunoglobulin antibody (Pierce Chemical, Rockford, IL) at 1:10,000 dilution for 1 hr. Between steps, membranes were extensively washed with Tris-buffered saline/Tween-20 three times in 30 min. The immunoreactive bands were visualized with the enhanced chemiluminescence detection system (Amersham, Buckinghamshire, UK). Absorbance of the bands were quantified by laser scanning densitometry (Amersham), and the amount of secreted form of βAPP was expressed as a percentage of control value in the same experiment.
For reverse transcription-PCR, total RNA was extracted from the cells by the guanidinium thiocyanate-acid-phenol method (24), and 1 μg of RNA was incubated with 100 pmol of random hexamer (Life Technologies, Gaithersburg, MD) at 70° for 10 min and reverse-transcribed by adding 4 μl of reaction buffer containing 20 mm Tris·Cl, pH 6.9, 90 mm KCl, 4.5 mm MgCl2, 150 μmβ-NAD, 10 mm(NH4)2SO4, 10 mm dithiothreitol, 0.5 mm dNTP mixture, and 10 units of reverse transcriptase, superscripts (10 units/μl; Life Technologies) at 37° for 40 min and 42° for 30 min. Resulting cDNA samples were added to 10 mm Tris-Cl, pH 9.0, 50 mm KCl, 0.1% Triton X-100, 3.5 mmMgCl2, 0.5 mm dNTP mixture, 10 pmol of primers, and 2.5 units of Taq DNA polymerase (5 units/μl; Poscochem, Sagnan, Korea). Primer sets to amplify βAPP cDNA (Amitof Biotech, Boston, MA) are 5′-CTACCACAACTACCACTGAG-3′ for 5′ primer, and 5′-TCATCTCCGGGGGTCTCCAG-3′ for 3′ primer, which correspond to the 824th to 843rd and 1135th to 1154th sequences of rat βAPP cDNA, respectively. Primer sets for β-actin (Amitof) are 5′-GATTACTGCTCTGGCTCCTA-3′ for 5′ primer, and 5′-CAGTAACAGTCCGCCTAGAA-3′ for 3′ primer. PCR reaction was performed for 30 cycles on Ericomp heating block, where each cycle consisted of denaturation at 94° for 1 min, primer annealing at 55° for 1 min, and extension at 72° for 2 min. The amplified products were subjected to separate on 8% PAGE and visualized by UV illumination in the presence of ethidium bromide. The ratios of the βAPP/β-actin signals were determined by densitometry.
Aβ was detected essentially as described previously (25). Briefly, PC12 cells were grown to near confluency in 100-mm dishes and transiently transfected with the vector containing Swedish type mutant βAPP695 (26) using liposome (LipofectAMINE; Life Technologies). After 24 hr, transfected cells were metabolically labeled for 16 hr with 50 μCi/ml [S35]methionine (1011 Ci/mmol; ICN, Cleveland, OH) in methionine-free Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum and 5% horse serum in the presence or absence of nicotine (100 μm). Conditioned media were immunoprecipitated with R1282 [against synthetic Aβ(1–40); 1:500 dilution] (25). Immunoprecipitated proteins were separated on 10–20% gradient Tris-Tricine SDS-PAGE.
For all findings, each condition was repeated in three to seven independent experiments. ANOVA followed by Duncan test, Wilcoxon rank sum test, or Student’s t test was used to determine the statistical significance.
Results
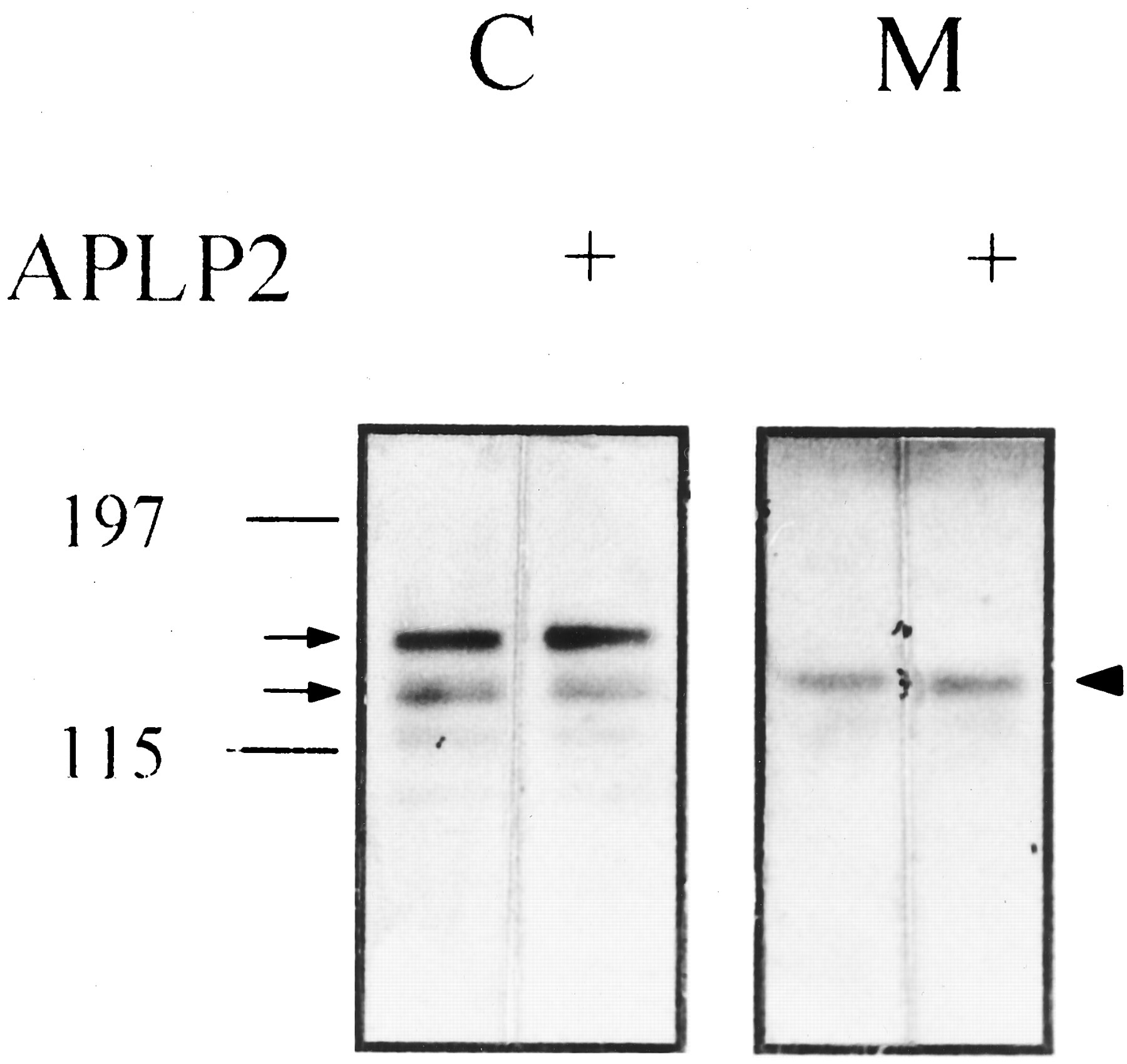
Because there have been some controversies with regard to the identity of the βAPP species detected in the conditioned medium of PC12 cells, we first characterized the βAPP derivatives in the medium with several specific antibodies. The 22C11, CT15, and C8 antibodies could all definitely detect the full-length βAPP species whose molecular mass ranged from 120 to 140 kDa in the lysates of PC12 cells (Fig. 1). According to the previous reports, the bands of 120 and 140 kDa are believed to represent immature (N′-glycosylated) and mature (N′- andO′ -glycosylated) form of βAPP, respectively. A 105-kDa band reactive with 22C11 might be a partially degraded product of βAPP that did not appear in other sets of samples (Fig. 2). In the conditioned medium, a protein band with apparent molecular mass of 130 kDa detected by 22C11 antibody was not reactive against CT15 and C8 antibodies, both of which were raised against the carboxyl-terminal portion of βAPP (Fig. 1). Thus this band in the conditioned medium of PC12 cells is not full-length but a carboxyl-terminally truncated form of βAPP, probably cleaved by α-secretase. 22C11 antibody was known to cross-react with APLP2. However, neither the 22C11 immunoreactive bands in the cell lysates nor that in the conditioned medium could be abolished by preadsorption with APLP2 protein, indicating that these species are all derived from βAPP (Fig.2). Next we examined the changes in the amount of βAPPs after nicotine treatment. As shown in Fig.3, nicotine increased the release of βAPPs in a concentration-dependent manner. The levels of βAPPs after treatment with 50 and 100 μm of nicotine were significantly different from that of the control group (p < 0.05 by ANOVA with Duncan test). The amount of βAPPs in the conditioned medium began to increase from 30 min after application of nicotine (100 μm), reached a maximal level at 3 hr, and tended to decrease thereafter (Fig.4). The maximal stimulation of βAPPs release by nicotine was 2.9-fold of basal level (Fig. 4B). The levels of βAPPs after 1- and 2-hr treatment with 100 μm of nicotine were significantly different from that of control group (p < 0.05 by ANOVA with Duncan). To determine whether the increase of βAPPs by nicotine was due to enhanced transcription of βAPP, we extracted total RNA from the cells and performed reverse transcription-PCR. However, there were no significant changes in the expression levels of three major isoforms of βAPP (βAPP695, βAPP751, and βAPP770) relative to β-actin from 30 min to 4 hr after nicotine treatment (Fig.5). Therefore the enhanced release of βAPPs by nicotine probably arises from an accelerated proteolytic processing rather than from an increased transcription of βAPP. Cotreatment of mecamylamine, a specific nicotinic receptor antagonist, significantly attenuated the release of βAPPs increased by nicotine (p < 0.05 by Wilcoxon rank sum test; Fig.6). Thus the effect of nicotine on βAPP processing was thought to be specifically mediated by nAChR. In addition, EGTA, a calcium chelator, almost completely abolished the enhancing effect of nicotine on βAPPs release (p < 0.01 by Student’s t test; Fig. 6), implying that Ca2+ entry through the nACh receptor is essential in the enhanced release of βAPPs by nicotine. Mecamylamine or EGTA itself had little effect on βAPP processing (data not shown). Then we examined whether the increase in βAPPs release by nicotine is accompanied by a decrease in the secretion of Aβ. To increase the amount of Aβ in the conditioned medium to an easily detectable level, we transiently transfected Swedish mutant βAPP695 to PC12 cells. However, nicotine (100 μm) treatment did not significantly change the amount of Aβ production in the transfected cells (data not shown).

Characterization of the βAPP derivatives in PC12 cells. Cell lysates (C; 90 μg) and conditioned medium (M; 25 μg) of PC12 cells were run on the same 5–14% gradient SDS-PAGE and immunoblotted with 22C11, CT15, or C8 antibody. The molecular mass of the prestained marker is shown at theleft. Arrows, bands representing mature and immature full-length βAPP (about 140 and 120 kDa, respectively);arrowhead, βAPPs (about 130 kDa).
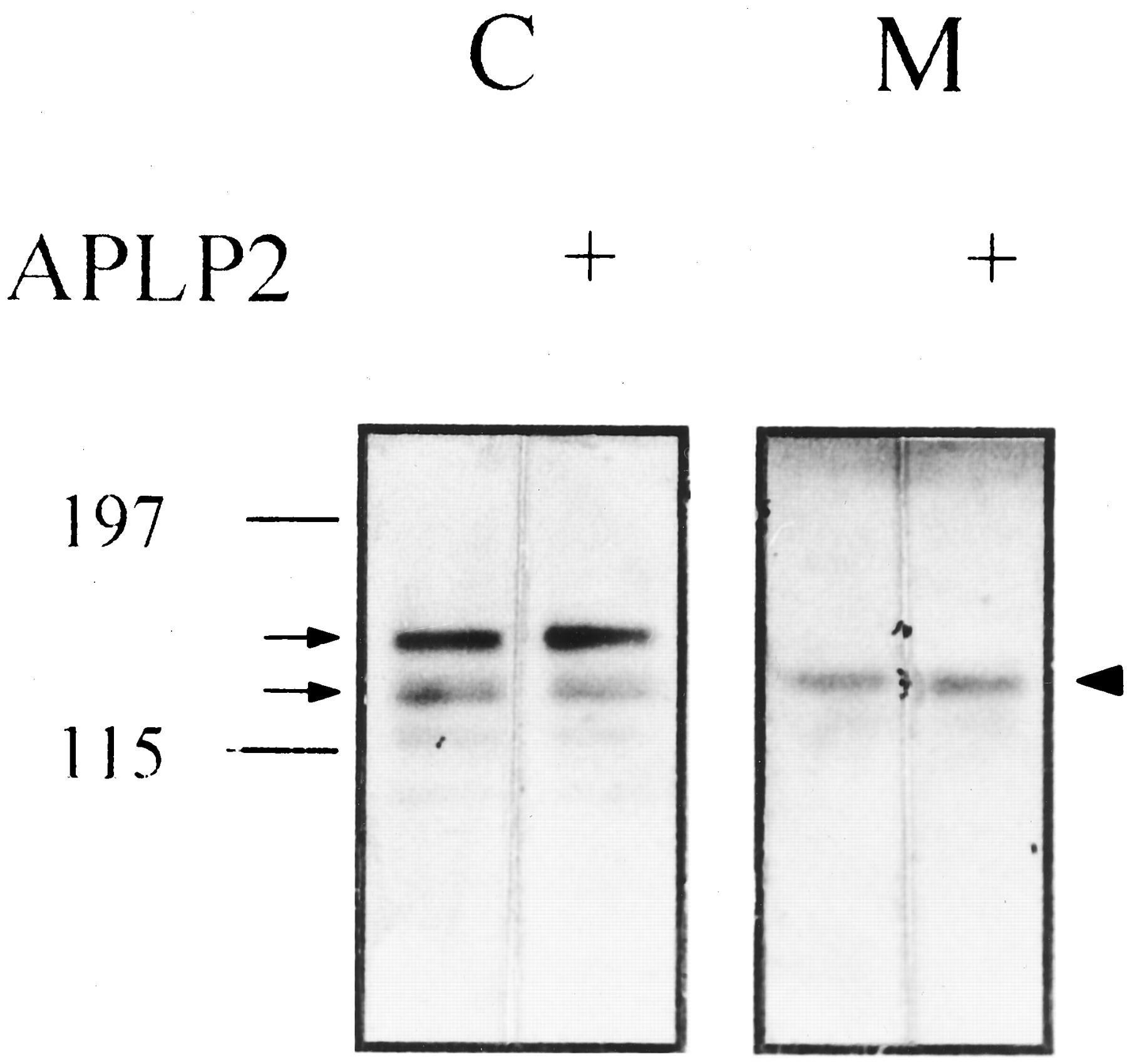
APLP2 preadsorption did not abolish the 22C11 immunoreactive bands in the cell lysates and that in the conditioned medium of PC12 cells. Cell lysates (C; 60 μg) and conditioned medium (M; 20 μg) of PC12 cells were run on the same 5–14% gradient SDS-PAGE and immunoblotted with 22C11 with or without preadsorption with APLP2 peptide (25 μg/ml). Left, molecular mass of the prestained marker. Arrows, bands representing mature and immature full-length βAPP in the cell lysates; arrowhead, βAPPs in the medium.

Concentration-dependent release of βAPPs from PC12 cells in response to nicotine. A, Representative immunoblot showing the effects of various concentrations (1, 10, 50, and 100 μm) of nicotine on the release of βAPPs in PC12 cells. After incubating the cells with the indicated concentration of nicotine for 4 hr, conditioned medium was collected and treated as described in Materials and Methods. The same amounts of extracellular proteins (15 μg) were run on 5–14% gradient SDS-PAGE and immunoblotted with 22C11 antibody. Left, molecular mass of the prestained marker. Arrowhead, band representing βAPPs (about 130 kDa) B, Quantitation of the βAPPs band in the blots. The amount of secreted form of βAPP was expressed as a percentage of the control value without nicotine treatment in the same experiment. Data are mean ± standard error values of three to seven different experiments. Bars, standard error. ∗, Significantly different from control value (p < 0.05) by ANOVA with Duncan test.

Time-dependent enhancing effect of nicotine on the secretion of βAPPs by nicotine. A, Representative immunoblot demonstrating the effect of nicotine (100 μm) on the production of βAPPs at various time points in PC12 cells. Conditioned media were analyzed as described in Fig. 3. Left, molecular mass of prestained marker; arrowhead, band representing βAPPs (about 130 kDa). B, Results of the densitometric measurements of the βAPPs band. The amount of βAPPs was expressed as a percentage of control value in the same experiment. Data are mean ± standard error values of three to five different experiments.Bars, standard error. ∗, Significantly different from control value (p < 0.05) by ANOVA with Duncan test.

Effects of nicotine treatment on the expression of βAPP mRNA in PC12 cells. After incubation with nicotine (100 μm) for the indicated time, total RNA was extracted from the cells, reverse transcribed, and amplified as described in Materials and Methods. The results of control (C) without any drug treatment and negative control (N.C.) in which PCR was done without reverse transcription are also shown. Arrowheads,bands representing βAPP770 (331 bp), βAPP751 (274 bp), βAPP695 (107 bp), and β-actin (120 bp); the size (bp) of the molecular mass marker (M) on the left is shown for comparison.

Blocking effects of mecamylamine, a nicotinic receptor antagonist, and EGTA on the enhancement of βAPPs release by nicotine in PC12 cells. After incubating the cells with nicotine alone (lane 1, 100 μm), in the presence of mecamylamine (lane 2, 10 μm), or EGTA (lane 3, 2.5 mm) for 4 hr, media were collected and analyzed as described in Fig. 3. The amount of secreted form of βAPP was expressed as a percentage of control value in the same experiment. Data are mean ± standard error values of four independent experiments. Bars, standard error. ∗, Significant difference from the nicotine-alone group (p < 0.05) by Wilcoxon rank sum test; ∗∗, Significant difference from the nicotine-alone group (p < 0.01) by Student’s t test.
Discussion
In this study we have examined the effects of nicotine on the expression and processing of βAPP. We employed the PC12 cell, a rat pheochromocytoma cell line, as a model system. It has been shown to constitutively express βAPP and contain functional nAChR (27). It expresses α3, α5, α7, β2, and β4 subunit isoforms that are similar to those in sympathetic ganglion (28). Treatment of PC12 cells with nicotine did not affect the levels of three major isoforms of βAPP mRNA at any time points examined but increased the release of βAPPs into the medium in a concentration (>50 μm)- and time (>2 hr)-dependent manner. The effect of nicotine on the secretion of βAPPs is attenuated by cotreatment with mecamylamine, a noncompetitive antagonist of nAChR, especially of a ganglionic type of nAChR. These results indicated nicotine could enhance the release of βAPPs through the specific interaction with nAChR.
Many groups have employed PC12 cells to study the regulation of βAPP processing by various agents and regarded the βAPP species found in the culture medium as a secreted form. However, Ripellino et al. (29) reported that the βAPP species in the conditioned medium of PC12 cells was immunoreactive with a polyclonal antiserum (R1) directed against the last 13 amino acids of βAPP. Recently, the same group further demonstrated that primary bovine chromaffin cells secrete full-length βAPP, which can be immunoprecipitated with R1, CT15, and C8 antibodies (30). Moreover, they showed high K+-induced depolarization and cholinergic receptor agonists including carbachol and nicotine stimulated the secretion of full-length βAPP. The secretion of βAPP was accompanied by other compounds of chromaffin granules such as catecholamines and chromogranin A. However, this finding is not reproducible in our system. In the present study, the βAPP derivative detected in the conditioned medium of PC12 cells was not reactive with C8 (22) and CT15 (23) antibodies, both of which have been well known to be immunoreactive with the cytoplasmic portion of βAPP. Another report demonstrating that βAPPs cleaved by α secretase could be detected in the intracellular vesicles of PC12 cells (31) supports our data indirectly. The explanation for these discrepancies is uncertain. Cell type difference (primary bovine chromaffin cells versus PC12 cells) and antibody specificity (R1 versus CT15 and C8 antibodies) might be partially responsible for the contradictory findings. It is also possible that both the full-length form and the carboxyl-terminally truncated secreted form of βAPP are released into the conditioned medium of PC12 cells. However, at least in our system, the major secreted species is believed to be the carboxyl-terminally truncated secreted form of βAPP, and nicotine enhances the release of this species. Because Efthimiopoulos et al. (30) employed metabolic labeling with [35S]methionine and an immunoprecipitation technique, which is much more sensitive than the immunoblot used in this study, they could possibly detect the small amounts of full-length βAPP that is not detectable by immunoblot.
The mechanism of enhancement of βAPPs release by nicotine is not clear at present. However, considering the fact that the effect of nicotine on βAPP processing was almost completely abolished by the calcium chelator, EGTA, it is probably related to calcium entry through nAChR. Neuronal nAChR channels have a greater Ca2+ permeability (PCa:PNa = 20 for α7 homomeric channel, 1–1.5 for other neuronal heteromeric channels) than muscle nAChR (PCa:PNa = 0.2) (15). In PC12 cells, PCa:PNa is approximately 2.5 (27). Calcium entry through the neuronal nAChR channel is sufficient to activate various Ca2+-dependent cellular processes (32), such as neurotransmitter release. Depolarization induced by nAChR stimulation further increases the Ca2+ influx through voltage-sensitive Ca2+ channels. Several studies have indicated that calcium can also regulate βAPP processing (13, 14, 33, 34). Buxbaum et al. (13) demonstrated that thapsigargin and cyclopiazonic acid, which inhibit intracellular Ca2+ uptake into the endoplasmic reticulum, increased βAPPs release in a PKC-independent manner. Furthermore, calcium ionophore A23187 was also shown to enhance the release of βAPPs in differentiated PC12 cells (14). Electrical depolarization, which also raises the intracellular Ca2+concentration, enhances the βAPPs release from the hippocampal slices (32). The exact molecular mechanism by which calcium modulates βAPP processing remains unclear. One possibility is that calcium-sensitive proteases might be directly involved in βAPP processing. The other possibility is that calcium might indirectly influence the activities of other proteases responsible for βAPP processing. Although little is known about the identity of α secretase(s), several proteases have been suggested as potential candidates. One of them is calcium-activated, dithiothreitol-sensitive metalloproteases present in rat brain (35). However, the exact identity of α secretase(s) and the role of Ca2+ in regulating the activity of the enzyme(s) needs to be further elucidated. It is of considerable interest that βAPPs can stabilize the intracellular Ca2+ concentration (3) by activating high conductance K+ channels. These results raise the possibility that βAPPs induced by increased intracellular Ca2+ may act as a negative regulator to control the intracellular level of Ca2+, an important signaling molecule in the neuron.
Although an increased release of βAPPs is expected to be accompanied by a decrease in Aβ secretion, this is not always true. Several studies demonstrated a dissociation between βAPPs release and Aβ generation (14, 34, 36). In the present study also, nicotine could not lower the Aβ production from the Swedish mutant βAPP transfectants, whereas it could stimulate the release of βAPPs. Thus there might be a complex regulatory mechanism for these two processing events of βAPP. However, because we examined the effects of nicotine only on the pathologically high production of Aβ, the modulation of the physiological Aβ production by nicotine needs to be established in future studies.
βAPPs has potent trophic and protective activities in several cell culture models. It can stimulate neurite outgrowth in PC12 cells, promote the proliferation of fibroblasts, and protect cultured neurons from metabolic and excitotoxic insults (3). Therefore βAPPs may act as a paracrine neurotrophic and neuroprotective factor. Interestingly, nicotine was also shown to attenuate the neuronal degeneration induced by glutamate (37) and nerve growth factor deprivation (38) in vitro. These findings were further extended to in vivostudies that demonstrated the protective effects of nicotine against neurotoxin-induced or mechanically induced degeneration of nigrostriatal dopaminergic neurons (39). Increased release of neurotrophic βAPPs by nicotine might partially explain the neuroprotective effects of nicotine.
Physiological relevance of the enhanced βAPPs release by nicotine is not clear. Nicotine has been shown to cause a myriad of psychopharmacological effects such as cognitive enhancement (16). The central effects of nicotine were believed to be principally mediated by neuronal nAChR. Activation of neuronal nAChR located in the presynaptic sites could facilitate the release of neurotransmitters, such as glutamate, γ-aminobutyric acid, and dopamine, and enhance synaptic transmission (40). Our data indicate that the processing of βAPP also can be modulated by nicotinic receptor activation. In AD brains, nicotinic neurotransmission is severely damaged (18), which may lead to an aberrant processing of βAPP. Reduced βAPPs release might secondarily contribute to the neuronal loss in AD.
Because the degree of cognitive impairments in AD has been reported to correlate well with the deficits of cholinergic neurotransmission in the brain (19), elevation of acetylcholine level was hypothesized to be helpful in improving the cognitive deficits in AD. Many groups have tried to supplement the cholinergic transmission by administration of acetylcholine precursors, muscarinic or nicotinic receptor agonists, or acetylcholinesterase inhibitors. Although most of them had failed to effectively ameliorate the symptoms of AD, pilot clinical studies indicated that nicotine might be beneficial for the treatment of the deficits in attention and information processing associated with AD (20, 21). However, nicotine itself has limited utility as a therapeutic agent because of its dose-limiting side effects such as hypertension, tachycardia, and abdominal pain. Thus many groups are now trying to develop a novel nicotinic receptor agonist that is able to enhance the cognitive functions by specific interaction with neuronal nAChR without eliciting peripheral side effects. Our results imply an additional benefit of nicotinic receptor agonists in AD because they not only directly supplement the nicotinic neurotransmission but also increase the secretion of neuroprotective βAPPs.
Acknowledgments
We thank Dr. Sangram S. Sisodia (Johns Hopkins University, Baltimore, MD) for kindly providing CT15 antibody, Dr. Dennis J. Selkoe (Brigham and Women’s Hospital, Boston, MA) for C8 and R1282 antibodies, Dr. Myung-Koo Lee (Chungbuk National University, Chongju, Korea) for PC12 cells, and Drs. Tae-Wan Kim and Robert Moir (Massachusetts General Hospital, Charlestown, MA) for purified APLP2 protein.
Footnotes
- Received November 15, 1996.
- Accepted May 27, 1997.
-
Send reprint requests to: Prof. Yoo-Hun Suh, Department of Pharmacology, College of Medicine, Seoul National University, 28 Yongon-dong, Chongno-gu, Seoul, 110–799 Korea. E-mail:yhsuh{at}plaza.snu.ac.kr
-
This study was supported by grants-in-aid from Korea Ginseng and Tobacco Research Institute (1994–1996) and Seoul National University Hospital (1997).
Abbreviations
- AD
- Alzheimer’s disease
- βAPP
- amyloid precursor protein
- βAPPs
- secreted form of amyloid precursor protein
- APLP2
- amyloid precursor-like protein 2
- Aβ
- amyloid β protein
- nAChR
- nicotinic acetylcholine receptor
- PAGE
- polyacrylamide gel electrophoresis
- PKC
- protein kinase C
- PLC
- phospholipase C
- PCR
- polymerase chain reaction
- ANOVA
- analysis of variance
- SDS
- sodium dodecyl sulfate
- bp
- base pair
- 5-HT
- 5-hydroxytryptamine
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}