

Abstract
Background: Pathological anxiety is the most common type of psychiatric disorder. The current first-line anti-anxiety treatment, selective serotonin/noradrenaline reuptake inhibitors, produces a delayed onset of action with modest therapeutic and substantial adverse effects, and long-term use of the fast-acting anti-anxiety benzodiazepines causes severe adverse effects. Inhibition of the fatty acid amide hydrolase (FAAH), the endocannabinoid N-arachidonoylethanolamine (AEA) degradative enzyme, produces anti-anxiety effects without substantial “unwanted effects” of cannabinoids, but its anti-anxiety mechanism is unclear.
Methods: We used behavioural, electro-physiological, morphological and mutagenesis strategies to assess the anti-anxiety mechanism of the FAAH inhibitors PF3845 and URB597.
Results: PF3845 exerts rapid and long-lasting anti-anxiety effects in mice exposed acutely to stress or chronically to the stress hormone corticosterone. PF3845-induced anti-anxiety effects and in vivo long-term depression (LTD) of synaptic strength at the prefrontal cortical input onto the basolateral amygdala neurons are abolished in mutant mice without CB1 cannabinoid receptors (CB1R) in brain astroglial cells, but are conserved in mice without CB1R in glutamatergic neurons. Blockade of glutamate N-methyl-d-aspartate receptors and of synaptic trafficking of glutamate AMPA receptors also abolishes PF3845-induced anti-anxiety effects in mice and LTD production in rats. URB597 produces similar anti-anxiety effects, which are abolished by blockade of LTD induction in mice.
Limitations: The determination of FAAH in which types of brain cells contribute to AEA degradation for the maintenance of amygdala interstitial AEA has yet to be determined.
Conclusion: We propose that the rapid anti-anxiety effects of FAAH inhibition are due to AEA activation of astroglial CB1R and subsequent basolateral amygdala LTD in vivo.
Introduction
Pathological anxiety, as manifested in anxiety disorders, is the most common type of psychiatric disorder, with a lifetime prevalence of 29%.1 The current first-line anti-anxiety medication, selective serotonin/noradrenaline reuptake inhibitors (SSRIs/SNRIs), produce a delayed onset of action2–4 with modest therapeutic effects and significant adverse effects.2,3 Benzodiazepines produce rapid anti-anxiety effects, but their long-term use causes severe adverse effects.5–7 It is therefore important to identify new fast-acting, long-lasting anti-anxiety medication with relatively mild adverse effects.
Endocannabinoid (eCB) signalling regulates synaptic plasticity.8 Anandamide (AEA) and 2-arachidonoylglycerol (2-AG) are 2 well-characterized eCBs, with their concentrations being principally regulated by the fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), the key enzymes to hydrolyze AEA and 2-AG, respectively.9,10 It has been reported that FAAH inhibitors exerted anti-anxiety effects11–14 without considerable “unwanted effects” of cannabinoids,15 while deletion of the FAAH gene prevented animals from experiencing anxiety-like behaviours.12,16 However, how FAAH inhibitors produce anti-anxiety effects remains unclear.
The amygdala is a key structure in the regulation of stress and anxiety responses. Acting as the primary interface for this regulation, the basolateral amygdala (BLA) receives glutamatergic input from the prefrontal cortex (PFC) and sends glutamatergic output to the central amygdala.17–19 Endocannabinoid signalling was found in synapses of all brain regions important for the processing of stress and anxiety, including the BLA.20,21 It is well established that eCBs are rapidly synthesized postsynaptically and then released into the synaptic clefts to activate presynaptic CB1R, suppressing presynaptic release of the excitatory neurotransmitter glutamate.8,15 Various lines of evidence led to the hypothesis that stress increases FAAH activity to reduce AEA concentrations, which increases the excitability of BLA principal neurons due to the unavailability of AEA for its suppression of glutamate release, leading to anxiety development.16,22,23 Therefore, FAAH inhibitors could produce anti-anxiety effects through decreased excitability of BLA principal neurons following AEA suppression of glutamate release.16,22,23 However, here we found results suggesting that FAAH inhibitors produce anti-anxiety effects through long-term depression (LTD) after sequential activation of astroglial CB1R and postsynaptic glutamate receptors at PFC–BLA synapses.
Methods
Animals
All procedures were performed in keeping with the guidelines established by the Canadian Council on Animal Care, as approved by the Animal Care Committee of the University of Ottawa Institute of Mental Health Research, which approved the present study (ACC-2012–004). Similar procedures were also approved by the Shaanxi Normal University, Xi’an, China. Animals were purchased from Charles River. Behavioural studies used male CD1 mice (30–35 g), male C57BL/6 mice (20–22 g) or male Sprague Dawley rats (250–300 g). Male Sprague Dawley rats were also used for patch clamp study (75–100 g) or in vivo electrophysiological study (250–300 g).
Similar to our recent study,24 CB1R-floxed mice were generated by Beijing Biocytogen Co., Ltd, China, and crossed with transgenic mice expressing the CreERT2 under control of the GFAP promoter (GFAP-CreERT2 mice) to obtain a GFAP-CB1R-knockout (KO) mouse line,25 or with the CaMKIIa-iCre transgenic mice expressing the improved Cre in brain projecting neurons to obtain a CaMKII-CB1R-KO mouse line.25 CB1R protein deletion from astroglial cells in GFAP-CB1R-KO mice and from glutamatergic neurons in CaMKII-CB1R-KO mice was confirmed in our recent study.25
Electrophysiological analysis on amygdala slices
Rats were decapitated 2 h after intraperitoneal administration of PF3845 (4 mg/kg) or vehicle or 45 min after intraperitoneal administration of N-arachidonoylserotonin (AA-5-HT; 5 mg/kg). Brains were removed and amygdala slices (400 μm) were prepared and maintained in an artificial cerebral spinal fluid (ACSF) solution. To record excitatory post-synaptic currents (EPSCs), BLA pyramidal cells were whole-cell voltage clamped at −70 mV, and patch pipettes were inserted into cells with extracellular circulation of ACSF. For depolarization-induced suppression of excitation (DSE), we depolarized cells from −70 to 0 mV for 5 s, and EPSCs were evoked at 4-s intervals. Some slices were perfused with AM281 (1 μM) before inducing DSE. We recorded spontaneous miniature EPSCs (mEPSCs) using a standard procedure with 1 μM TTX and 10 μM bicuculline in the perfusion solution. We collected EPSCs by voltage clamp recordings using a stimulus pulse consisting of a single square wave (stimulation intensity: 100 μs, 50–200 μA).
To record BLA LTD in brain slices, adult mutant mice were decapitated 2 h after intraperitoneal injection of vehicle or PF3845 (4 mg/kg) for preparation of coronal vibrosections (300 μm) of the amygdala slices. We identified BLA projection neurons according to their characteristics.26 The field excitatory postsynaptic potential (fEPSP) was evoked at 0.05 Hz with an electrode placed approximately 500 μm from the recorded neuron, close to the fibre tract of the external capsule adjacent to the BLA. Stimulus pulse intensities were 0.3–1.5 mA with a duration of 100 μs. We induced LTD with low-frequency stimulation (LFS) at 1 Hz for 15 min.
Electrophysiological analysis in anesthetized rats
As described,24,27 under anesthesia with 40% urethane (3 mL/kg administered intraperitoneally) and 4% chloral hydrate (3 mL/kg administered intraperitoneally) rats received insertion of a stimulating electrode into the PFC (AP +3.0, ML 0.7, D/V −4.6 mm) and a recording electrode into the BLA (AP −2.7, ML 4.9, D/V −7.2 mm). The fEPSP was evoked by stimulation with a square-wave constant current pulse of 0.033 Hz and 0.2 ms duration. After baseline recording for 20 min, rats received the following treatment (administered intraperitoneally unless otherwise stated) before a continuous recording of fEPSPs for 2 h: PF3845 (4 mg/kg), URB597 (0.6 mg/kg) or vehicle; AEA (50 pmol/0.5 μL, intra-BLA) or vehicle; AM281 (2 mg/kg) or vehicle 20 min before or after PF3845; actinomycin-D (72 μg/12 μL administered by intracerebroventricular injection) or vehicle 30 min before PF3845; AA-5-HT (5 mg/kg) or vehicle; Ro25–6981 (6 mg/kg) or NVP-AAM077 (1.2 mg/kg) 20 min before PF3845; or the LTD-blocking peptide Tat-GluR2 (3 μmol/kg) or its scrambled sequence Tat-GluR2s24,25 1 h before PF3845.
Mouse surgery
With isoflurane anesthesia, mice received implantation of 2 guide cannulas (22 Ga) into the bilateral BLA (AP −1.35, ML ± 3.1, DV −5.0 mm), followed by affixing the cannulas to the skull. One week later, intra-BLA injection of Tat-GluR2 or Tat-GluR2s with 0.5 μL for each side (30 pmol/per injection, 0.1 μL/min) was performed, followed 30 min later by intra-peritoneal PF3845 injection (4 mg/kg), with the behavioural test conducted 2 h later. Finally, mice were perfused with 4% paraformaldehyde, and brains were cut to confirm cannula placement.
Behavioural tests
To receive acute inescapable swimming stress, a mouse was placed into a Plexiglas cylinder (65 × 30 cm) filled with 25°C water to a height of 10 cm for 5, 10 or 20 min. The mouse then underwent a standard open field test (OFT) and elevated plus maze test (EPMT) in a dimly lit room 1 h later. To mimic the important feature of chronic stress, animals were given corticosterone (CORT) solution (25 μg/mL, free-base) or vehicle as drinking water for 21 days, with an average CORT exposure of 6.5 mg/kg/mouse.25,28 One week after CORT exposure, mice underwent the OFT and EPMT in sequence on the same day. Immediately after the EPMT, mice were deprived of food but had free access to water for 24 h before undergoing the novelty-suppressed feeding test (NSFT), during which each rodent was placed in a corner of a novel chamber for 6 min, with a food pellet on the centre floor. We measured the time taken by the mouse to explore the novel environment to the point when it was sitting on its haunches and eating food with its forepaws. Then we measured 5-min food consumption in the mouse home cage. A delayed non-matching to sample task (DNMTST) was conducted as described in our recent study.24 Briefly, Sprague Dawley rats were restricted food intake to maintain their body weight at about 85% of their normal weight. Spatial working memory (SWM) was examined in a wooden T-shaped box for the DNMTST, consisting of pretest training, acquisition training and performance of the test.24
Animals received the following treatment administered intraperitoneally unless otherwise stated: PF3845 (4 mg/kg) or vehicle 2 h before acute stress, followed 1 h later by the OFT and EPMT; AM281 (2 mg/kg), MK801 (0.1 mg/kg), NVP-AAM077 (1.2 mg/kg), Ro25–6981 (6 mg/kg) or vehicle 10 min before PF3845, followed 2 h later by acute stress with a behavioural test conducted 1 h later; Tat-GluR2 or Tat-GluR2s (3 μmol/kg) 30 min before PF3845 or URB597 (0.6 mg/kg), followed 2 h later by acute stress with a behavioural test conducted 1 h later; CORT (20, 40 or 80 mg/kg) 1 h before a behavioural test; RU486 (20 mg/kg administered subcutaneously) 30 min before acute stress, followed 1 h later by a behavioural test. Mutant mice received PF3845 (4 mg/kg) or vehicle 2 h before acute stress, followed 1 h later by a behavioural test; for the DNMTST, HU210 (0.05 mg/kg), PF3845 (4 or 8 mg/kg) or vehicle was given 30 min or 2 h before the first trial in each testing day. For chronic CORT-exposed mice, PF3845 (4 or 8 mg/kg) or vehicle was injected once or once every other day for 8 days, with or without AM281 (2 mg/kg), vehicle or Tat-GluR2 pretreatment 10 min or 1 h before PF3845, followed 2 h, 24 h or 1 week later by a behavioural test.
Morphological analysis
After behavioural testing, mice exposed to chronic CORT or vehicle with or without 1 or subchronic injection of PF3845 (4 or 8 mg/kg administered intraperitoneally) were decapitated under anesthesia. Brains were removed and processed for rapid Golgi staining following manufacturer’s instructions (FD Neurotech). Golgi-stained tissue was sliced (120 μm) and mounted, and 4–5 spiny pyramidal-like neurons were reconstructed per animal, as this class of BLA neurons exhibits architectural changes in response to stress.16 We used the following criteria to choose cells:16 presence of untruncated dendrites, cell bodies existing within the BLA boundaries, isolation of the cell body from neighbouring impregnated neurons and a clear relationship of the primary dendrite to the soma. We used custom-designed macros embedded in NIH ImageJ software for morphometric analysis of digitized images, including quantification of the total dendritic length and total number of branch points along the dendritic tree.
Statistical analysis
Results are reported as means ± standard errors of the mean (SEM). We performed statistical analyses of the data using a Student t test, 1-way analysis of variance (ANOVA) or 2-way ANOVA, followed by a least significant difference (LSD) post hoc test. We considered results to be significant at p < 0.05.
Detailed information on the statistical method and results is provided in Appendix 1, Table S1, available at jpn.ca.
Results
PF3845 does not significantly affect presynaptic release of glutamate
PF3845 shows exceptional potency and selectivity to FAAH,10 as 1–10 mg/kg completely blocked FAAH activity to produce maximal elevations in brain AEA levels.29 We conducted major experiments with an intraperitoneal injection of 4 mg/kg of PF3845. In agreement with a recent study,30 recording of BLA pyramidal cells from naive rats revealed induction of DSE, which was abolished by bath application of AM281 onto amygdala slices (Fig. 1A and B), suggesting mediation of BLA DSE by eCB activation of presynatic CB1R. However, DSE remained unchanged in brain slices from PF3845-treated rats (Fig. 1A and B). PF3845 exposure in vivo reduced mEPSC amplitude without significant effects on mEPSC frequency (Fig. 1C and E). Similar results were observed after injection of the dual FAAH inhibitor and TRPV1 receptor antagonist AA-5-HT (Fig. 1C and E),31 although AEA induces synaptic LTD through activation of postsynaptic TRPV1 receptors.32–35 These results together indicate that PF3845 does not significantly affect presynaptic release of glutamate.
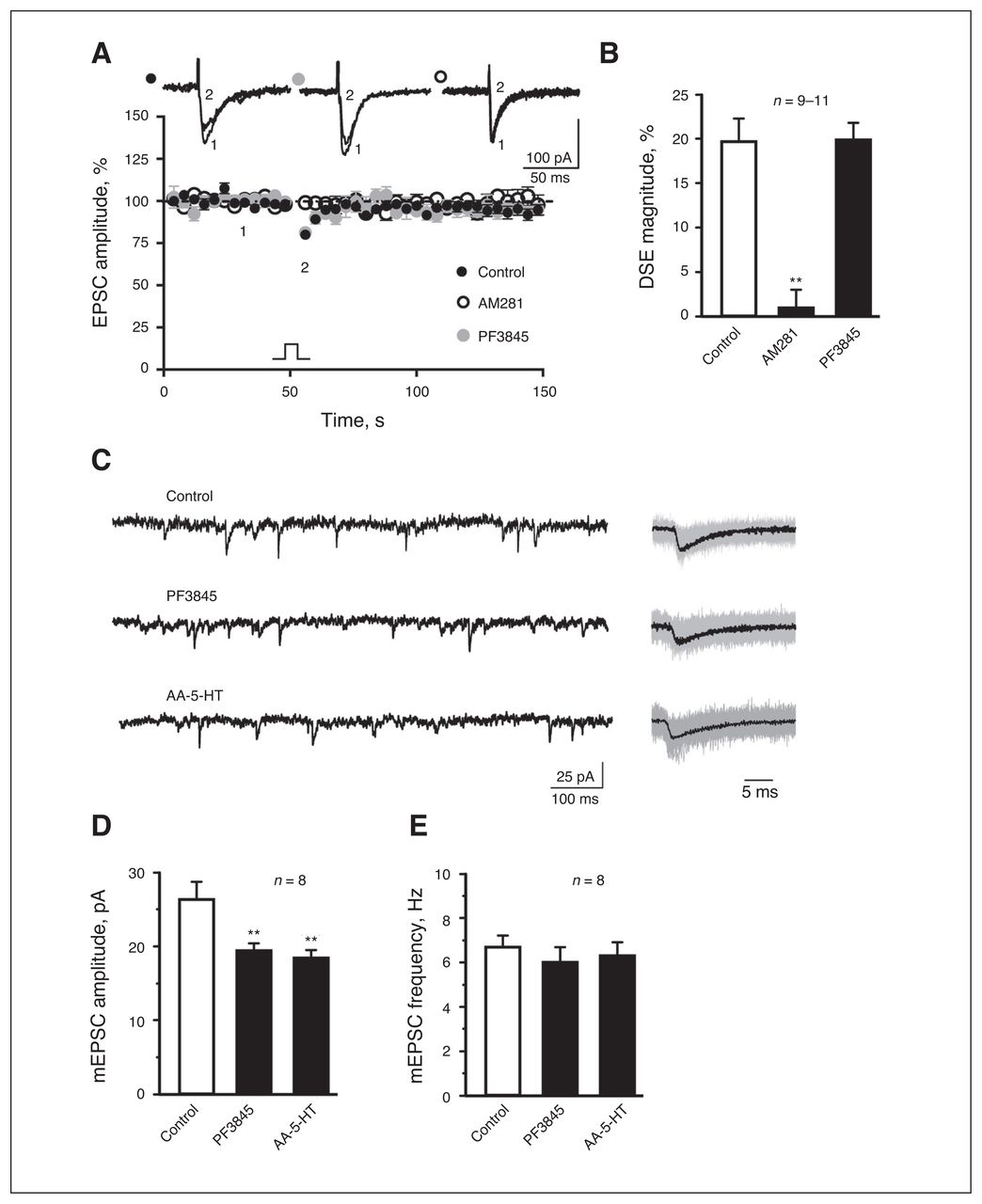
PF3845 does not significantly affect presynaptic release of glutamate in rat slice preparations. (A) A plot of normalized excitatory postsynaptic current (EPSC) amplitude and (B) summary histogram show that bath application of AM281 (1 μM), but not PF3845 exposure in vivo (4 mg/kg administered intraperitoneally), significantly decreases depolarization-induced suppression of excitation magnitude. (A) Representative EPSC traces are superimposed on the top of the plot. (C–E) Representative mEPSC traces (C: left, 1 s; right, 30 ms averaged) and (D, E) summary histograms show that PF3845 (4 mg/kg administered intraperitoneally) and AA-5HT (5 mg/kg administered intraperitoneally) significantly reduce mEPSC amplitude without significant effects on mEPSC frequency. All summary graphs show means ± standard errors of the mean. **p < 0.01 versus control, least significant difference post hoc test after 1-way analysis of variance (B: F2,27 = 21.66, p < 0.01; D: F2,21 = 7.007, p < 0.01; E: F2,21 = 0.327, p = 0.72). DSE = depolarization-induced suppression of excitation.
FAAH inhibition induced LTD at PFC-BLA synapses via astroglial CB1R
The fEPSP amplitudes at PFC–BLA synapses in anesthetized rats decreased by approximately 20% from baseline at 2 h after an intraperitoneal injection of PF3845 or URB597 (Fig. 2A and H). PF3845 application elevated brain AEA levels for more than 24 h,29 but PF3845-elicited synaptic depression for more than 2 h is LTD rather than multiple transient synaptic depressions for 3 reasons. First, while LTD maintenance, but not transient synaptic transmission depression, requires new protein synthesis,36 injection of the RNA transcription inhibitor actinomycin-D37 before PF3845 blocked the late-but not the early-phase expression of LTD (Fig. 2B and H). Second, an intra-BLA application of AEA also induced in vivo LTD at PFC–BLA synapses (Appendix 1, Fig. S1A and S1B). Third, AM281 injection 20 min before but not 20 min after PF3845 injection blocked LTD induction (Fig. 2C and H). Notably, AA-5-HT (5 mg/kg administered intraperitoneally)31 also induced in vivo LTD at PFC–BLA synapses (Appendix 1, Fig. S1C and S1D).

Fatty acid amide hydrolase (FAAH) inhibitors induce in vivo long-term depression (LTD) at prefrontal cortex–basolateral amygdala (PFC–BLA) synapses in rodents. (A–C, F, G) Plots of normalized field excitatory postsynaptic potential (fEPSP) amplitude in anesthetized rats show that an administration of vehicle, PF3845 (PF) or URB597 at 0 min elicits LTD lasting longer than 2 h (A). Although actinomycin-D blocks the late-phase expression of PF3845-elicited LTD (B), PF3845-elicited LTD is prevented by pretreatment with AM281 (C), Ro25,6981 (Ro in F) and Tat-GluR2 (G), but not posttreatment with AM281 (C) or pretreatment with NVP-AAM077 (NVP in F) or Tat-GluR2s (G). (D, E) Plots of normalized EPSP amplitude in mouse brain slices show that low-frequency stimulation (LFS) induces LTD in brain slices prepared from GFAP-CB1R-knockout (KO) mice but not from GFAP-CB1R-wild-type (WT) littermates with an in vivo PF3845 injection (D) and that LFS induces in vitro LTD from CaMKII-CB1R-KO mice with an in vivo vehicle injection (Ca-CB1R-KO-Veh) but not from CaMKII-CB1R-WT (Ca-CB1R-WT) and CaMKII-CB1R-KO mice (Ca-CB1R-KO) with an in vivo PF3845 injection (E). Representative fEPSP traces before (1) and after (2) vehicle, PF3845 or URB597 administration (A–C, F, G) or LFS (D, E) are shown below each plot. (H) Average percent change of fEPSP amplitude before (1) and after (2) vehicle, PF3845 or URB597 administration or LFS as depicted in panels A–G. Both A and F share the same PF3845 group. All summary graphs show means ± standard errors of the mean. **p < 0.01 versus vehicle, WT mice or PF3845, Student t test (B–D, G) or least significant difference post hoc test after 1-way analysis of variance (A: F2,12 = 28.995, p < 0.01; E: F2,24 = 23.301, p < 0.01; F: F2,12 = 28.787, p < 0.01).
Because it is extremely difficult to record in vivo fEPSPs from BLA in anesthetized mice, we used an indirect strategy to measure in vivo LTD. It is known that in vivo LTD in living mice would occlude in vitro LTD induction at the same synapses in brain slices prepared from the same mice. LFS induced in vitro LTD at PFC–BLA synapses in brain slices prepared 2 h after PF3845 injection from GFAP-CB1R-KO but not from GFAP-CB1R wild-type (WT) littermates (Fig. 2D and H). Notably, LFS also induced in vitro LTD in slices prepared from CaMKII-CB1R-KO mice with an intraperitoneal injection of vehicle but not from CaMKII-CB1R-WT and CaMKII-CB1R-KO mice with an intraperitoneal injection of PF3845 (Fig. 2E and H). Thus, PF3845 could induce in vivo LTD at PFC–BLA synapses from GFAP-CB1R-WT, CaMKII-CB1R-WT and CaMKII-CB1R-KO mice but not from GFAP-CB1R-KO mice. We next studied whether PF3845-induced in vivo LTD at PFC–BLA synapses was affected by NVP-AAM077 and Ro25–6981, antagonists of NR2A- and NR2B-containing N-methyl-d-aspartic acid receptor (NR2AR and NR2BR), respectively,24 and by Tat-GluR2 peptide, which selectively interrupts endocytosis of postsynaptic α-amino-3-hydroxy-5-methyl-isoxazolepropionic acid receptors (AMPAR).24 Ro25–6981 and Tat-GluR2, but not NVP-AAM077 or Tat-GluR2s, blocked PF3845-induced LTD at PFC–BLA synapses (Fig. 2G and H). Thus, FAAH inhibition induces in vivo LTD at PFC–BLA synapses likely via activation of astroglial CB1R and neuronal NR2BR and AMPAR internalization in the BLA.
Anti-anxiety effects of FAAH inhibition on acute stress-exposed mice through LTD
Stress plays a key role in the etiology and/or development of anxiety disorders.38 We found that an acute stress for 10 or 20 min, but not 5 min, enhanced anxiety-like behaviours of naive mice in the EPMT and OFT (Appendix 1, Fig. S2A and S2B). In the following experiments, we applied 10-min acute stress to naive mice.
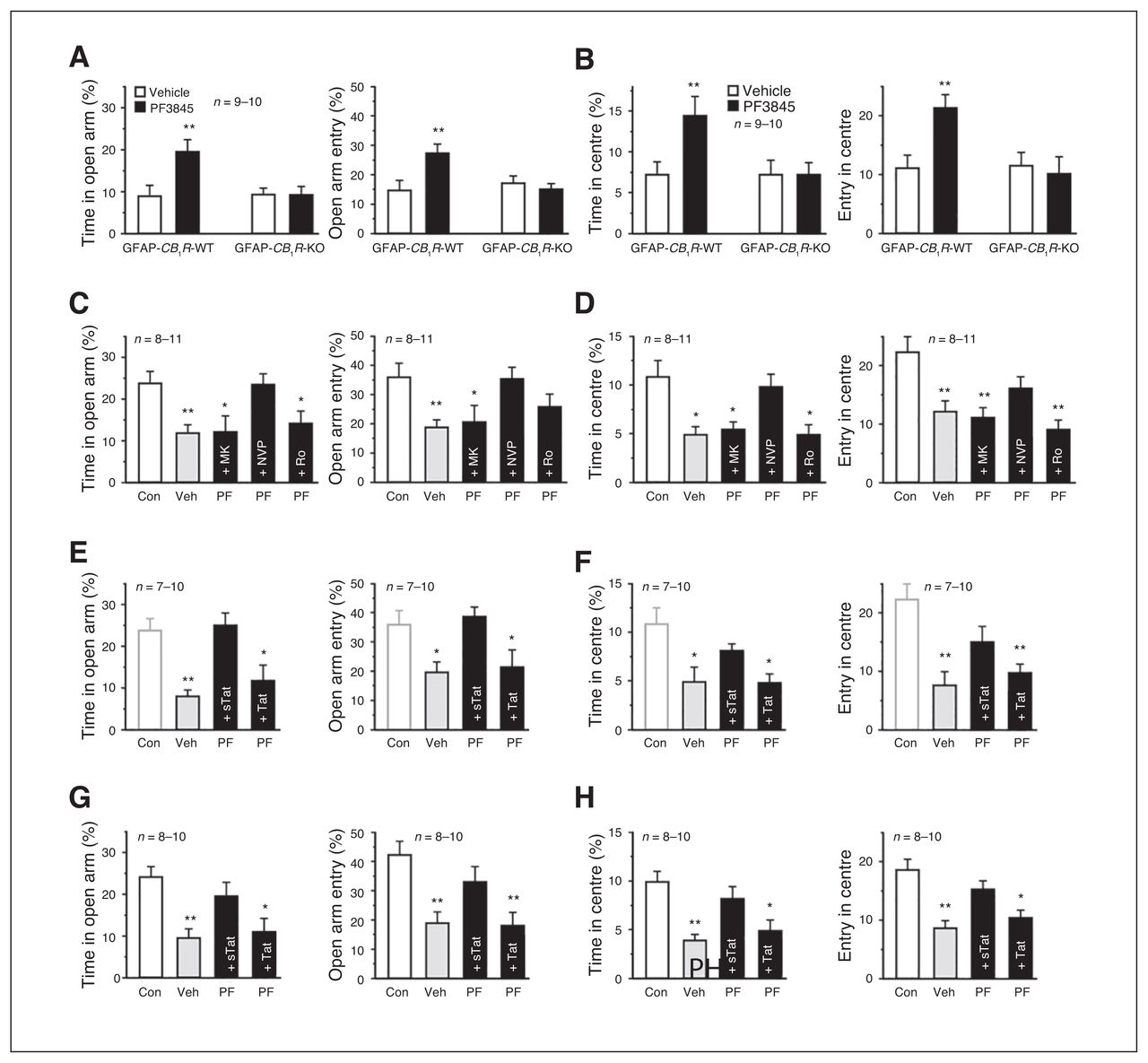
PF3845 blocked acute stress-elicited anxiogenic effects in the EPMT and OFT, which was abolished by AM281 pretreatment (Appendix 1, Fig. S3A and S3B), indicating that AEA activation of CB1R is responsible for anti-anxiety effects produced by PF3845. Relative to vehicle, which did not produce significant differences between WT and mutant littermates, PF3845 exerted significant anti-anxiety effects in GFAP-CB1R-WT mice, but not in GFAP-CB1R-KO littermates, in both the EPMT (Fig. 3A) and OFT (Fig. 3B). Neither vehicle nor PF3845 exerted significant differences between CaMKII-CB1R-WT and CaMKII-CB1R-KO littermates, with significant anti-anxiety effects elicited by PF3845 (Appendix 1, Fig. S3C and D). The NMDAR antagonist MK801 and Ro25–6981, but not NVP-AAM077, abrogated PF3845-induced anti-anxiety effects in both the EPMT and OFT (Fig. 3C and D). Both systemic and intra-BLA injection of Tat-GluR2, but not Tat-GluR2s, abolished anti-anxiety effects of both PF3845 and URB597 in both the EPMT and OFT (Fig. 3E–H and Appendix 1, Fig. S3E and F). Thus, the anti-anxiety effects of PF3845 on acute stress-exposed mice likely require activation of astroglial CB1R and neuronal NR2BR and AMPAR internalization in the BLA.

PF3845 exerts anti-anxiety effects in acutely stressed mice via astroglial-mediated long-term depression (LTD). (A, B) Relative to vehicle, PF3845 increased time spent in and entrance times into both the elevated plus maze test (EPMT) open arms (A) and open field test (OFT) centre area (B) in GFAP-CB1R-wild type (WT) but not GFAP-CB1R-knockout (KO) litter-mates. (C–H) Relative to control mice without stress (Con), acutely stressed mice receiving vehicle (Veh) show anxiogenic effects. Both an intraperitoneal injection (C–F) and intra–basolateral amygdala (BLA) application (G, H) of MK801 (MK), Ro25–6981 (Ro; C, D) and Tat-GluR2 (Tat; E–H), but not NVP-AAM077 (NVP) or Tat-GluR2s (sTat), abolish the behavioural actions of PF3845 (PF) in EPMT open arms (C, E, G) and OFT centre area (D, F, H). The Con in E and F are the same as Con in C and D. All summary graphs show means ± standard errors of the mean. *p < 0.05 and **p < 0.01 versus vehicle or control, least significant difference post hoc test after 2-way analysis of variance (ANOVA; A: left, F1,35 = 4.929, p < 0.05; right, F1,35 = 6.793, p < 0.05; B: left, F1,35 = 3.653, p = 0.06; right, F1,35 = 5.533, p < 0.05) or after 1-way ANOVA (C: left, F4,40 = 4.328, p < 0.01; right, F4,40 = 3.347, p < 0.05; D: left, F4,40 = 5.83, p < 0.01; right, F4,40 = 6.247, p < 0.01; E: left, F3,31 = 7.295, p < 0.01; right, F3,31 = 4.175, p < 0.05; F: left, F3,31 = 5.979, p < 0.01; right, F3,31 = 9.939, p < 0.01; G: left, F3,33 = 6.747, p < 0.01; right, F3,33 = 6.853, p = 0.01; H: left, F3,33 = 8.877, p < 0.01; right, F3,33 = 9.928, p < 0.01).
Rapid and long-lasting anti-anxiety effects of PF3845 on chronic CORT-exposed mice through LTD
CORT and its antagonist RU486 respectively mimicked and blocked the anxiogenic effects of acute stress (Appendix 1, Fig. S2C–F), suggesting a key role of CORT in mediating acute stress-elicited anxiogenic effects. Thus, we examined whether PF3845 exerted anti-anxiety effects in mice receiving CORT for 3 weeks.39,40 Chronic CORT-exposed mice showed significant anxiety behaviours in the EPMT, OFT and NSFT (Appendix 1, Fig. S2G–I). Subchronic injection of PF3845 (4 mg/kg, 4 injections administered once every 2 d) produced significant anti-anxiety effects in the EPMT, OFT and NSFT, which were blocked by AM281 pretreatment (Fig. 4A–F). Two hours after injection, 8 but not 4 mg/kg PF3845 exerted rapid anti-anxiety effects in the EPMT (Fig. 4G and H) and NSFT (Fig. 4K and L), but not in the OFT (Fig. 4I and J), which was blocked by Tat-GluR2. One week after injection, PF3845 (8 mg/kg administered intraperitoneally) still exerted anti-anxiety effects in the EPMT (Fig. 4M and N) and the NSFT (Fig. 4Q and R), but not in the OFT (Fig. 4O and P). Thus, both CB1R activation and AMPAR internalization are necessary for PF3845 to produce rapid anti-anxiety effects on chronic CORT-exposed mice.

PF3845 produces anti-anxiety effects in mice chronically exposed to corticosterone (CORT) for 3 weeks. (A–F) Relative to vehicle injection (Veh), subchronic injection of PF3845 (PF; 4 mg/kg administered intraperitoneally) increases time spent in and entrance times into both the elevated plus maze test (EPMT) open arms (A, B) and open field test (OFT) centre area (C, D), reduces latency to eat in a novel environment (E) without significant effects on food intake in home cages (F), all of which are reversed by AM281 (AM) pretreatment (A–F). (G–L) Relative to vehicle injection (Veh), 8 but not 4 mg/kg of PF3845 (PF8 and PF4, respectively) at 2 h after injection increases time spent in and entrance times into the EPMT open arms (G, H) and reduces latency to eat in a novel environment (K), all of which are reversed by Tat-GluR2 pretreatment (Tat). Neither 8 nor 4 mg/kg of PF3845 significantly affects time spent in and entrance times into the OFT centre area (I, J) and food intake in home cages (L). (M–R) Relative to vehicle injection (Veh), mice at 1 week after a PF3845 (PF) injection (8 mg/kg administered intraperitoneally) show increased time spent in and entrance times into the EPMT open arms (M, N) and reduced latency to eat in a novel environment (Q) without significant changes on time spent in and entrance times into the OFT centre area (O, P) and food intake in home cages (R). All summary graphs show means ± standard errors of the mean. *p < 0.05 and **p < 0.01 versus vehicle, least significant difference post hoc test after 1-way analysis of variance (A: F2,29 = 19.14, p < 0.01; B: F2,29 = 8.68, p < 0.01; C: F2,29 = 4.265, p < 0.05; D: F2,29 = 5.530, p < 0.01; E: F2,27 = 3.455, p < 0.05; F: F2,27 = 0.177, p = 0.84; G: F3,40 = 3.601, p < 0.05; H: F3,40 = 3.395, p < 0.05; I: F3,40 = 5.863, p < 0.01; J: F3,40 = 2.408, p = 0.081; K: F3,39 = 3.146, p < 0.05; L: F3,39 = 0.17, p = 0.92) or Student t test (M–R).
Recent studies showed an association of increased anxiety behaviours with increased dendritic arborization, complexity or spine density of BLA pyramidal neurons.16,41–43 Relative to control mice, chronic CORT-exposed mice showed a significant increase in both dendritic length and branching points of BLA pyramidal neurons, which were reversed 1 week after a PF3845 injection (8 mg/kg administered intraperitoneally) or 1 day after subchronic PF3845 injection (4 mg/kg, 4 injections administered once every 2 d; Fig. 5).

PF3845 reverses basolateral amygdala (BLA) structural remodelling by chronic corticosterone (CORT) exposure in mice. (A–C) Representative images (A) and summary histograms (B, C) show that relative to control (Con), chronic CORT exposure in vivo increases dendritic length and branching points of the dendritic trees of the BLA pyramidal neurons, which are completely reversed by subchronic treatment of PF3845 (CORT + 4 × PF) or a single treatment of PF3845 (CORT + 1 × PF). (A) Bar = 50 μm. All summary graphs show means ± standard errors of the mean. **p < 0.01 versus control, least significant difference post hoc test after 1-way analysis of variance (B: F3,111 = 9.083, p < 0.01; C: F3,111 = 13.658, p < 0.01).
Absence of PF3845 effects on working memory
We recently showed impairment of working memory by synthetic cannabinoid HU210.24 After acquisition of the SWM task with more than 80% correct choices (Appendix 1, Fig. S4A), rats received a test session either 30 min after injection of HU210 or 4 or 8 mg/kg of PF3845 or 2 h after 4 or 8 mg/kg of PF3845. HU210, but not 4 or 8 mg/kg of PF3845, impaired SWM performance (Appendix 1, Fig. S4B), suggesting that PF3845, with its anti-anxiety dosage in both acute stress and chronic stress hormone-treated animals, does not significantly impair SWM.
Discussion
Here, we investigated anti-anxiety behavioural responses of the selective and potent FAAH inhibitors PF3845 and URB597 in stress-exposed animals. We observed that PF3845 exerts rapid anti-anxiety effects in mice exposed acutely to stress or chronically to CORT as well as long-lasting anti-anxiety effects at 1 week after a single PF3845 administration or at 1 day after its subchronic injection for 8 days in chronic CORT-exposed mice. Interestingly, these anti-anxiety responses were associated with reversible dendrite remodelling of BLA pyramidal neurons. In the context of anti-anxiety effects produced by FAAH inhibition, our results expand the recent findings that the FAAH inhibitor URB597 produced anti-anxiety effects 1 day after acute stress exposure14 and that subchronic injection of PF3845 exerted anti-anxiety effects in chronic unpredictable mild stress–exposed animals.44 FAAH inhibitors did not produce many unwanted effects elicited by marijuana or cannabinoids, including hypomotility, physical dependence, tolerance and loss of CB1R function.15,45–47 We showed here that PF3845 did not significantly impair SWM, while a major side effect of marijuana is working memory impairment in humans48 and animals.24 All these lines of evidence together support the potential therapeutic value of FAAH inhibitors to treat stress-related anxiety disorders in humans. This is important especially considering that the current first-line anti-anxiety medication with SSRIs/SNRIs produces a delayed onset of action of 2–12 weeks with modest therapeutic effects in up to 69% of patients and significant adverse effects (e.g., nausea, sexual dysfunction) in up to 49% patients,2–4 whereas the long-term use of rapid anti-anxiety medication with benzodiazepines causes severe adverse effects, including dependence, withdrawal syndromes,5 risk of dementia6 and traffic accidents.7
Genetic deletion of the FAAH gene or chronic exposure of FAAH inhibitors during chronic restraint stress prevented animals from developing anxiety-like behaviours.16,22,23 Humans and mice homozygous at the FAAHA/A allele, which leads to a destabilized FAAH enzyme and an increased AEA signalling, show anti-anxiety behavioural responses with increased functional connectivity of the PFC–BLA.49 These findings suggest a key role of increased FAAH activity and, consequently, decreased AEA signalling in the development mechanisms of anxiety disorders. Indeed, chronic stress increased the hydrolytic activity of amygdala FAAH, reduced amygdala contents of AEA, and increased dendritic arborization, complexity or spine density of BLA pyramidal neurons.16 Chronic stress-induced decrease of AEA signalling and subsequent increase of presynaptic neurotransmitter release likely occur at glutamatergic PFC–BLA synapses, because chronic stress increased the intrinsic excitability, sensitivity to afferent stimulation and activation of BLA pyramidal neurons.50,51 The increased excitability and sensitization is likely mediated by structural remodelling of BLA pyramidal neurons as chronic stress increases dendritic arborization, induces new spine formation and facilitates the formation of excitatory inputs to BLA pyramidal neurons.16,52 Indeed, BLA pyramidal neurons express FAAH,53 whereas pre-synaptic CB1R was shown in brain slices to modulate excitatory glutamatergic inputs onto BLA pyramidal cells.54 Therefore, many researchers have recently hypothesized that stress increases FAAH activity to reduce AEA signalling, which increases excitability of BLA principal neurons due to the unavailability of AEA for the suppression of glutamate release, leading to anxiety-like behaviour.16,22,23 Accordingly, they proposed that FAAH inhibition could reduce presynaptic glutamate release onto BLA glutamatergic pyramidal neurons through an increase of AEA and subsequent activation of CB1R, leading to anti-anxiety responses.16,22,23 However, here we provide evidence strongly supporting an alternative mechanism underlying anti-anxiety effects of FAAH inhibitors.
Endocannabinoid activation of glutamatergic presynaptic CB1R can induce either short-term synaptic depression (i.e., DSE or LTD), depending on its pattern of synthesis.8 Because FAAH inhibition was hypothesized to reduce presynaptic glutamate release onto glutamatergic BLA pyramidal neurons,16,22,23 we examined whether PF3845 exposure in vivo could reduce presynaptic glutamate release, leading to decreased DSE at BLA glutamatergic synapses in brain slices. However, we did not detect significant effects of PF3845 on DSE. Nevertheless, these results are not conclusive because BLA DSE may be specifically associated with 2-AG but not AEA signalling.30 Our further studies using mEPSC recordings suggest that PF3845 exposure in vivo does not significantly affect presynaptic release of glutamate. AEA can also activate postsynaptic TRPV1 receptors to induce AMPAR endocytosis, leading to postsynaptically expressed LTD.31–34 However, PF3845-increased AEA is unlikely to activate postsynaptic TRPV1 receptors to induce postsynaptically expressed LTD, because further experiments with the dual FAAH inhibitor and TRPV1 receptor antagonist AA-5-HT suggest that PF3845 activates postsynaptic glutamatergic receptors without significant effects on postsynaptic TRPV1 receptors at PFC–BLA synapses.
Next, we observed from anesthetized rats that PF3845 exposure in vivo induced in vivo LTD at PFC–BLA synapses that requires CB1R but not TRPV1 receptors. Interestingly, our electrophysiological experiments with mutant mice lacking the CB1R gene selectively in brain astroglial cells or glutamatergic/GABAergic projecting neurons suggest the requirement of CB1R in astroglial cells but not glutamatergic neurons for PF3845-increased AEA to induce in vivo LTD at PFC-BLA synapses.
CB1R is located in GABAergic and glutamatergic presynaptic terminals8,20,21 and in astroglial cells24 having a highly ramified morphology and occupying more than 50% of brain volume.55 With their unique morphology and position, astroglial cells are fairly exposed to brain interstitial fluid and synapses.56 Our findings of astroglial CB1R24 indicate that the interstitial space contains eCB that can directly stimulate astroglial CB1R. This idea is supported by in vivo microdialysis studies showing consistent baseline levels of interstitial AEA in various brain regions.57–59 A systemic injection of the FAAH inhibitor URB597 or PF3845 significantly increased interstitial AEA but not 2-AG levels,57–59 and mutant mice without the FAAH gene showed a 2-fold increase of interstitial AEA levels.58 These data support the notion that amygdala interstitial AEA forms a basal AEA stream (similar to hippocampal interstitial 2-AG stream25) with a constant supply and a continuous clearance by FAAH. Thus, an acute inhibition of FAAH would produce an acute accumulation of interstitial AEA, leading to astroglial CB1R activation.
Our further experiments demonstrated the requirement for NR2BR activation and postsynaptic AMPAR endocytosis for PF3845 to induce postsynaptically expressed LTD in vivo at glutamatergic PFC–BLA synapses. Our recent study also showed the requirement of astroglial CB1R, NR2BR and post-synaptic AMPAR for synthetic cannabinoids and MAGL inhibitors to induce in vivo LTD at hippocampal CA3-CA1 synapses.24,25 Thus, the signalling pathway mediating FAAH inhibitor-induced in vivo LTD at PFC–BLA synapses mirrors that mediating synthetic cannabinoid- and MAGL inhibitor–induced in vivo LTD at hippocampal CA3-CA1 synapses.24,25 Interestingly, PF3845-induced anti-anxiety effects were abolished both in mutant mice lacking the CB1R gene in astroglial cells and by pretreatment with either NR2BR antagonist or AMPAR endocytosis-blocking peptide. Thus, the anti-anxiety effects of PF3845 likely require activation of astroglial CB1R and neuronal NR2BR and subsequent AMPAR internalization in the BLA.
Limitations
FAAH is highly expressed not only in rat glutamatergic neurons in many brain regions, including the BLA,53 but also in cultured astroglial and microglial cells.60,61 The identification of FAAH in which types of these brain cells contribute to AEA degradation for the maintenance of interstitial AEA stream has yet to be determined.
Conclusion
We showed that a novel form of AEA-induced long-term synaptic plasticity in the BLA appears to mechanistically underlie anti-anxiety effects of FAAH inhibition in vivo. Our findings are consistent with a scenario, depicted in Figure 6, in which increased interstitial AEA stream by FAAH inhibition in vivo stimulates astroglial CB1R to increase ambient glutamate, which in turn activates NR2BR to trigger AMPAR internalization at PFC–BLA synapses. These events ultimately induce in vivo LTD at PFC–BLA synapses, suppressing BLA pyramidal neurons to produce anti-anxiety effects.

Proposed model for anti-anxiety effects of fatty acid amide hydrolase (FAAH) inhibition. FAAH inhibition increases basolateral amygdala (BLA) interstitial levels of N-arachidonoylethanolamine (AEA) to activate astroglial CB1R, which promotes the accumulation of interstitial glutamate to produce postsynaptic NR2BR activation, AMPAR endocytosis and long-term depression (LTD) expression, leading to anti-anxiety effects. Thus, FAAH inhibitors produce rapid anti-anxiety effects on acutely and chronically stressed mice through astroglial CB1R-mediated LTD at prefrontal cortex (PFC)–BLA synapses.
Acknowledgements
This project was supported by operating grants from the Canadian Institutes of Health Research to X. Zhang (MOP123249, MOP123256), a general grant from the National Natural Science Foundation of China to J. Han (grant no. 31371137), and an equipment grant from the Canadian Foundation for Innovation to X. Zhang. Portions of these data were included in the Innovations Award Lecture presented by X. Zhang during the CCNP meeting in Toronto, Canada, in May 2013.
Footnotes
↵* Share first authorship;
↵† share senior authorship.
Competing interests: None declared.
Contributors: All authors designed the study. T. Duan, N. Gu, Y. Wang and F. Wang acquired the data, which all authors analyzed. T. Duan, N. Gu, Y. Yang, Y. Shen, J. Han and X. Zhang wrote the article, which all authors reviewed and approved for publication.
- Received June 20, 2016.
- Revision received August 26, 2016.
- Revision received October 4, 2016.
- Revision received October 12, 2016.
- Revision received October 25, 2016.
- Revision received October 30, 2016.
- Accepted November 1, 2016.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools





