
Abstract
Despite robust evidence for the heritability of schizophrenia, postmortem studies have not traditionally linked cellular and molecular neuropathology with underlying genetic mechanisms in this disorder. The completion of the first draft of the Human Genome Project and the use of novel strategies in studying complex genetic disorders including schizophrenia have led to the identification of a growing list of schizophrenia susceptibility genes. In this review, we describe the strategy used to incorporate 2 potential schizophrenia susceptibility genes in the postmortem investigation of the pathophysiology of schizophrenia driven by 2 well-established hypotheses, the dopamine hypothesis and the neurodevelopmental hypothesis. The first gene codes for catechol-O-methyltransferase, an enzyme involved in catecholamine degradation, and the second gene codes for brain-derived neurotrophic factor, a growth factor implicated in cell survival, synaptogenesis and the development of cortical pyramidal neurons.
Introduction
Schizophrenia is a neuropsychiatric disorder characterized by hallucinations, delusions, thought disorder, deficit symptoms and cognitive dysfunction, with symptoms typically manifesting themselves in adolescence and early adulthood.1 Studies of the pathophysiology of this disorder have focused on the heritability of schizophrenia, the affected neurotransmitter systems and neuroanatomical abnormalities. A number of relatively consistent findings have emerged from these approaches. First, twin concordance rates, adoption studies, and genetic linkage and association analyses strongly suggest that schizophrenia has a heritable component.2,3 Second, the neurotransmitters dopamine (DA) and glutamate have been implicated through pharmacologic interventions that either mimic or exacerbate (N-methyl-d-aspartate [NMDA] receptor antagonists) or reduce (DA receptor antagonists) psychotic symptoms of schizophrenia. 4–7 Third, structural abnormalities in the brain include increased ventricular size along with reduced brain volume in the dorsolateral prefrontal cortex (DLPFC) and the entorhinal cortex, hippocampus and thalamus8–12 of patients with schizophrenia. The function of the DLPFC is also affected in schizophrenia, as demonstrated by decreased activation during the performance of cognitive tasks that are impaired in this disorder.13,14
Postmortem studies of schizophrenia have been hampered by a host of confounding variables. Although it is possible to match experimental subjects on the basis of gender, ethnic origin and postmortem interval (PMI), or time since death, other variables are more difficult to control. These include agonal state, ambient temperature after death and the accurate calculation of PMI. Moreover, the assumption that patients with schizophrenia and healthy controls can be matched ignores the higher incidence of alcohol and substance abuse in patients with schizophrenia, the consequences of a lifetime of treatment with neuroleptics and other drugs, the stress of acute exacerbations of illness and admissions to hospital, and the effects of a chronic mental illness on quality of life.15 On the other hand, postmortem studies allow a level of resolution not yet available through the use of imaging approaches and are invaluable in elucidating the pathophysiology of this disorder.
The completion of the first draft of the Human Genome Project followed by the discovery of allelic variants has presented schizophrenia researchers with an opportunity to link heredity to neurochemistry and neuroanatomy. It also allows genotyping of subjects with schizophrenia and controls, so that we can further elucidate the role of genes in systems of interest and refine the design of postmortem studies. Several strategies have been used in the study of the role of genes in schizophrenia. One approach is a top-down strategy, initially targeting large chromosomal loci implicated in schizophrenia by linkage analyses,16,17 with subsequent study of single nucleotide polymorphisms (SNPs) or groups of SNPs (haplotypes) within the implicated region. These SNPs and haplotypes are then examined in clinical association studies to determine whether they are associated with schizophrenia itself or intermediate phenotypes of the disorder. If so, then a gene can presumably be identified that contains the associated SNP or haplotype. Currently, at least 8 genes have been identified as schizophrenia susceptibility genes. Several of the genes under investigation as schizophrenia susceptibility genes are involved in glutamatergic signalling (G72, DAAO, RGS4 and NRG1),18 providing a link between hypotheses generated from imaging and neuropathologic studies and modern genetic approaches. Another gene implicated in schizophrenia by linkage analysis and involved in neurotransmission, the α–7 nicotinic acetylcholine receptor subunit gene (CHRNA7), has been identified as a schizophrenia susceptibility gene based on polymorphisms in the promoter region that appear more frequently in patients with schizophrenia as compared with unaffected controls. 19 In addition to genes involved in neurotransmission, a growing number of genes implicated in synapse formation, maintenance and plasticity have emerged as schizophrenia susceptibility genes. This list includes the aforementioned NRG1,20 as well as DTNBP121 and brain-derived neurotrophic factor (BDNF).22,23 Surely the list of schizophrenia susceptibility genes will continue to grow; the question is, however, how can we understand the functional implications of a susceptibility gene product on neurobiology in order to elucidate the pathophysiology of schizophrenia and eventually develop successful treatments? In this manuscript, we will describe the strategy we have used in the study of 2 genes in an attempt to link genetic vulnerability to the neurobiology of schizophrenia. The first gene codes for a DA-metabolizing enzyme catechol-O-methyltransferase (COMT), which was recently added to the list of schizophrenia susceptibility genes based on the discovery of an association between a haplotype of COMT and schizophrenia.24 A common SNP in this DA-metabolizing enzyme alters enzymatic activity and has functional implications for both cortical function and dopaminergic neurotransmission. The second gene examined is the gene for BDNF. At present, the status of the BDNF gene as a schizophrenia susceptibility gene is controversial; however, BDNF is an important neurotrophic factor for cortical glutamatergic pyramidal neurons and may play a role in synaptic pathology observed in the syndrome. More importantly, BDNF expression is known to be altered in schizophrenia25–27 and, therefore, if it is not itself a susceptibility gene, it is likely to be a downstream target of such a gene.
COMT
The COMT gene provides an excellent example of the use of new genetic information to understand an old hypothesis (the DA hypothesis of schizophrenia). The protein product of the COMT gene is an enzyme that catabolizes DA. Recently, COMT has been shown to contain a functional polymorphism resulting from a Val→Met substitution at the 108/158 locus in the peptide sequence. The Val allele substitution increases the efficiency of the enzyme 4-fold in comparison with the Met allele.28 On the basis of differential enzymatic activity, Val/Val individuals are expected to have decreased synaptic DA levels in the prefrontal cortex (PFC), Met/Met individuals to have high DA levels and Val/Met individuals to have intermediate DA levels. In imaging studies of healthy controls, this difference in COMT genotype has been shown to affect PFC function29–32 during working memory and other cognitive tasks known to depend on PFC DA levels. Patients with schizophrenia perform poorly on these cognitive tasks,33–35 and their PFC is not normally activated during performance of these tasks.33,35–38 These functional abnormalities have been related to cortical DA activity in in-vivo studies, 36,39–41 and the dopaminergic innervation of the PFC in schizophrenia is reduced.42 Last, inheriting a COMT Val allele has been found in association studies to increase the risk for schizophrenia slightly,29,43–45 implicating COMT as a susceptibility gene for schizophrenia. Thus, this COMT polymorphism, a susceptibility gene for schizophrenia, alters performance in healthy controls on a task that is known to depend upon DA levels in the PFC and is impaired in this disorder.
Dopaminergic signalling in schizophrenia is altered not only in the PFC but in subcortical structures such as the striatum as well. Dopaminergic tone in the PFC has indirect downstream effects on mesencephalic DA neurons. 46–48 A possible explanation for the relation between PFC excitatory cortical neurons and mesencephalic DA neurons has emerged from rodent experiments.49 In this proposed mechanism, prefrontal neurons tonically inhibit striatal DA projections, presumably through γ-aminobutyric acid (GABA)-ergic interneurons (Fig. 1).48,50,51 This model predicts that decreased PFC DA tone will result in a lessening of indirect tonic inhibition of mesencephalic DA neurons and, consequently, DA release from the mesencephalic neurons projecting to the striatum. Animal studies have substantiated this model,52,53 which provides a parsimonious explanation for the coexistence of cortical hypodopaminergia and subcortical hyperdopaminergia in schizophrenia.36,54,55
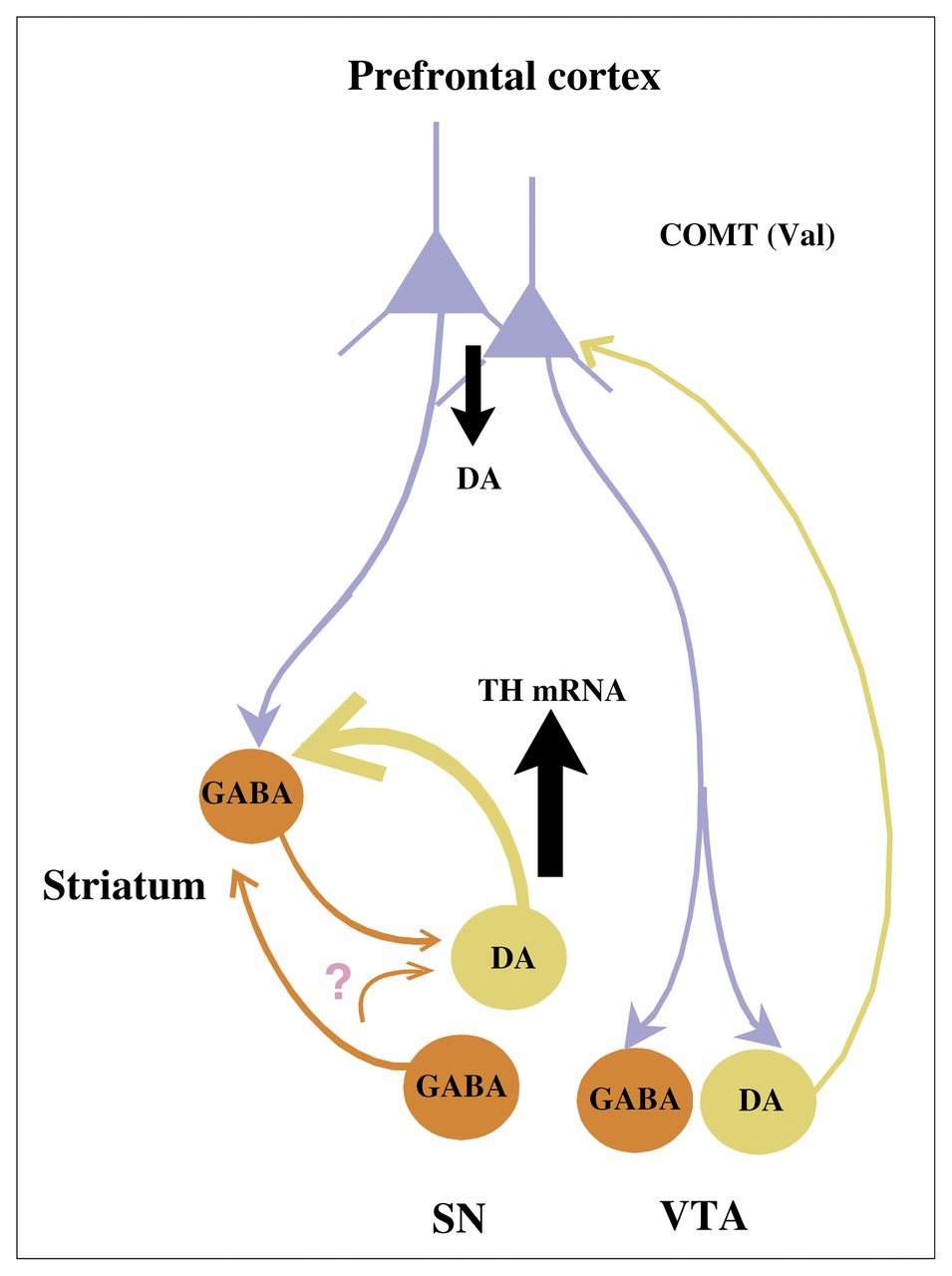
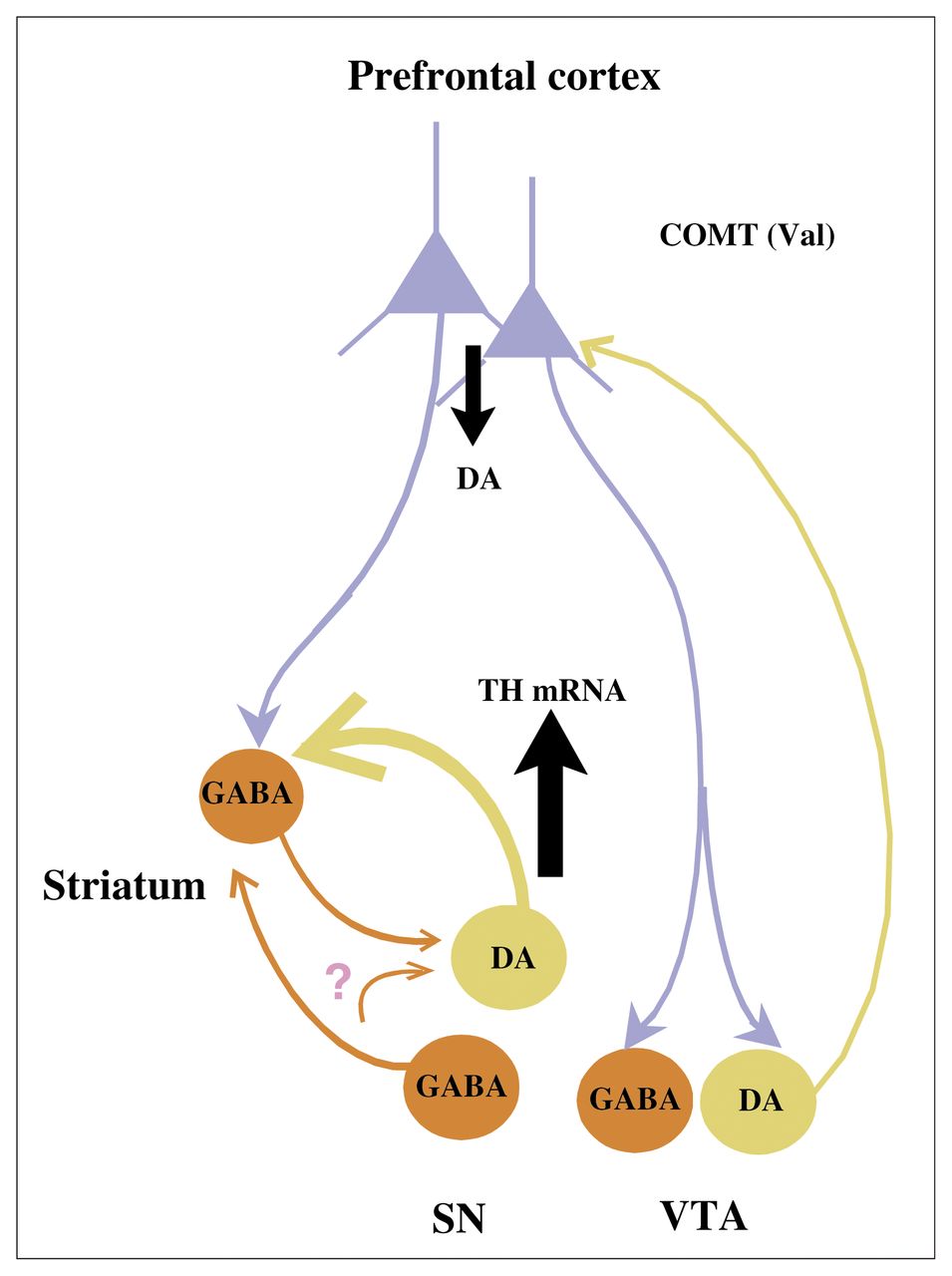{kind=link}
Diagram of proposed circuitry and its role in the effects of the catechol-O-methyltransferase (COMT) genotype on tyrosine hydroxylase (TH) gene expression in the brain stem. We predict that the Val/Val genotype of the COMT enzyme leads to reduced dopamine (DA) levels in the prefrontal cortex (PFC) relative to the Val/Met genotype and that indirect PFC projections via γ-aminobutyric acid (GABA) neurons in the striatum or mesencephalon lead to increased gene expression of TH mRNA in DA cell groups projecting subcortically. Some of the GABA projections remain to be confirmed; see question mark. SN = substantia nigra, VTA = ventral tegmental area. Figure adapted with permission from the Society for Neuroscience (J Neurosci 2003;23:2008–13).51
If this COMT polymorphism alters DA levels in the PFC, does it also influence subcortical DA? One would predict that inheritance of a Val allele, which is associated with increased COMT enzymatic activity, presumably leading to decreased DA levels in the PFC, would result in relatively increased recruitment of mesencephalic DA activity. To test this hypothesis, using in situ hybridization, we examined the expression of mRNA for tyrosine hydroxylase (TH), the rate-limiting enzyme for DA biosynthesis, in 5 mesencephalic DA cell groups in postmortem human brain specimens from healthy subjects carrying the Val/Val or the Val/Met genotypes.51 In addition, we examined the expression of dopamine transporter (DAT) mRNA and cyclophilin mRNA as controls for DA-related and DA-unrelated genes, respectively, in the same cell groups. A significant main effect of genotype on TH mRNA levels was found in the dorsal and ventral tiers of the substantia nigra pars compacta (SND and SNV, respectively). There was no effect of genotype on DAT or cyclophilin mRNA expression. The SNV and SND in nonhuman primates project to the striatum and amygdala, 46 suggesting that the effects of the COMT genotype on TH regulation are greatest in cell groups that do not project back to the PFC. Our results are consistent with the theoretical model outlined earlier and suggest a mechanism by which the COMT Val allele increases risk for schizophrenia (decreased DA signalling in PFC and increased DA signalling subcortically).
In summary, we were able to use postmortem studies to elucidate some of the biologic effects of COMT polymorphisms on the circuitry involved in schizophrenia.
BDNF
The gene for BDNF has been reported to be a schizophrenia susceptibility gene,22,23 and some association studies suggest that inheritance of certain BDNF alleles may relate to age of onset of the disease, responsiveness to treatment and parietal lobe volume in patients with schizophrenia.56,57 However, no clear association with the clinical diagnosis of schizophrenia was found in a number of studies.56–61 It is important to note that further study of genetic association of allelic variation in the BDNF gene with schizophrenia is required before a determination can be made as to whether or not BDNF is a schizophrenia susceptibility gene.
Reductions in BDNF mRNA25–27 and protein27 in the PFC of patients with schizophrenia, which cannot be readily explained by allelic variation in the BDNF gene itself, have been recently described in separate cohorts. These data suggest that the BDNF gene may be an important downstream target of one or more schizophrenia susceptibility genes. Therefore, future studies of the role of BDNF in schizophrenia should explore the relation between the genotypes of the schizophrenia susceptibility gene(s) under investigation and BDNF expression levels. Similarly, other genes shown to display reliable alterations in mRNA or protein expression in patients with schizophrenia should be examined as potential targets of schizophrenia susceptibility genes.
As previously described, one of the prevailing hypotheses concerning the pathophysiology of schizophrenia involves abnormalities in glutamatergic signalling. Patients with schizophrenia demonstrate cortical glutamate dysfunction as evidenced by a reduction in both glutamate and a neurochemical marker of glutamatergic neuronal integrity, N-acetyl-aspartate.62–65 Recent evidence points to synaptic pathology as a possible component of glutamatergic dysfunction in the cortex of patients with schizophrenia. Glutamatergic neurons in the DLPFC of patients with schizophrenia show a reduction of mRNAs encoding presynaptic proteins,66–68 as well as a reduction in synapse-associated proteins.69–72 Both the density of dendritic spines in layer III pyramidal neurons73,74 and the cortical neuropil are reduced in the DLPFC of patients with schizophrenia, the latter relating to increased neuronal density and decreased soma size of pyramidal neurons.75–78 Upregulation of BDNF increases both neuronal size and synaptic density, making it an excellent candidate for investigation in schizophrenia.79
BDNF is synthesized by neurons in the rodent frontal cortex80–84 and by pyramidal neurons in the DLPFC of primates, including humans.27,85,86 This neurotrophin is a trophic factor for glutamatergic neurons, as evidenced by increases in cell survival in vitro87–89 and stimulation of the growth of dendrites and increases in spine density of glutamatergic pyramidal neurons in the neocortex.90,91 In addition to synthesis and release in response to afferent activity, BDNF also modulates synaptic density and long-term potentiation of glutamatergic cortical neurons.89–95 Last, BDNF is critical for the formation of excitatory synapses, as evidenced by in-vivo temporal contiguity between increases in cortical BDNF mRNA and cortical neuron dendrite growth and synapse formation.96–102
We recently tested the hypothesis that glutamate-related pathology in the brain of patients with schizophrenia is associated with abnormal BDNF expression in the DLPFC.27 Using quantitative Western blotting, RNase protection assays and in situ hybridization, we detected a reduction in both BDNF protein and mRNA in postmortem specimens from patients with schizophrenia as compared with matched healthy controls. BDNF mRNA was localized to pyramidal neurons throughout layers II, III, V and VI, with patients with schizophrenia showing a reduction in BDNF expression in layers III, V and VI, suggesting that these neurons may provide less trophic support to their targets. In support of the mRNA data, quantitative Western blotting revealed a 40% reduction in BDNF protein in patients with schizophrenia as compared with healthy controls. With careful attention to experimental design, we were able to test for the effects of age, PMI, neuroleptic treatment history and history of depression, all of which may influence BDNF mRNA levels.84,101,103–109 Because BDNF exerts potent effects on forebrain systems believed to be involved in schizophrenia, our results further our understanding of glutamatergic dysfunction in schizophrenia and provide new avenues of research in synaptic pathology. Our results, coupled with the possible association of the BDNF gene with schizophrenia, raise the important question of how inheritance of certain forms of the BDNF gene may affect BDNF gene expression or BDNF functions within the DLPFC and how this, in turn, may confer increased risk for schizophrenia.
Conclusion
The addition of a genetic component to the existing neuropathologic approach to understanding the pathophysiology of schizophrenia has far-reaching implications. We now have the tools required to subtype experimental subjects genetically, thereby eliminating a heretofore uncontrolled-for confounding variable inherent in postmortem studies. By understanding which allelic variants of genes such as COMT are more often associated with schizophrenia, experiments can focus on specific gene products and, potentially, on interacting gene products or biologic pathways. In this way, information can be generated implicating specific proteins, cell populations, neural circuits and neuroanatomical structures altered in schizophrenia. The identification of these genetic susceptibilities may ultimately lead to clinical interventions with an emphasis on developing more effective treatments for this debilitating disorder.
Footnotes
Medical subject headings: brain-derived neurotrophic factor; catechol-O-methyltransferase; dopamine; genetic predisposition to disease; glutamate; prefrontal cortex; schizophrenia; substantia nigra; tyrosine hydroxylase.
Competing interests: None declared.
- Received July 4, 2003.
- Revision received February 4, 2004.
- Accepted February 17, 2004.