

Abstract
Objective: To explore the possible involvement of second-messenger pathways in the pathophysiology of bipolar disorder and the mechanism of action of mood stabilizers, we investigated the effects of dextroamphetamine (a model for mania) and the most widely used mood stabilizers, lithium chloride, sodium valproate and carbamazepine, on intraplatelet levels of calcium ion ([Ca2+]).
Design: In the first part of the study, dextroamphetamine was administered in vivo in a double-blind, placebo-controlled, crossover design. In the second part of the study, platelets from untreated subjects were incubated in vitro with dextroamphetamine, lithium chloride, sodium valproate or carbamazepine.
Participants: Fifteen healthy men between 18 and 45 years of age.
Outcome measures: Basal, thrombin-induced and serotonin- (5-HT) induced intraplatelet [Ca2+] determined by means of fura-2 fluorescent intensity.
Results: In vivo administration of dextroamphetamine had no effect on basal or agonist-induced intraplatelet [Ca2+]. However, in vitro basal platelet [Ca2+] was significantly higher in samples incubated with dextroamphetamine (86.8 nmol/L [standard error of the mean, SEM, 3.9], p < 0.001), lithium chloride (76.4 nmol/L [SEM 3.1], p < 0.002), sodium valproate (82.7 nmol/L [SEM 3.7], p < 0.001) and carbamazepine (84.8 nmol/L [SEM 3.3], p < 0.001) than in the controls (58.2 nmol/L [SEM 2.3]). Thrombin-induced and 5-HT-induced peak cytosolic [Ca2+] were significantly greater than control levels in samples incubated with carbamazepine (277.1 nmol/L [SEM 19.9] v. 195.8 nmol/L [SEM 12.2], p < 0.002; and 153.0 nmol/L [SEM 8.2] v. 115.4 nmol/L [SEM 5.7], p < 0.003, respectively).
Conclusions: This study does not support the involvement of intraplatelet [Ca2+] in the dextroamphetamine model of mania; however, the modulation of intraplatelet [Ca2+] by the mood stabilizers lithium chloride, sodium valproate and carbamazepine implicates intracellular [Ca2+] in the therapeutic mechanisms of these drugs and the pathophysiological basis of mania.
Introduction
Bipolar disorder, a psychiatric condition characterized by mood swings between mania and depression, occurs in approximately 1% of the general population.1 Pharmacotherapy with mood stabilizers remains the most effective treatment for long-term symptom reduction. Second-messenger pathways have been implicated in the development and neuronal pathophysiology of bipolar disorder. They are also therapeutic targets of mood stabilizers, but their mechanisms remain elusive. In particular, the phosphatidyl inositol (PI) cycle has been extensively investigated since the proposal of the inositol-depletion hypothesis.2,3 According to this hypothesis, the widely used mood stabilizer lithium may exert its therapeutic effects by inhibition of PI-cycle signalling. Subsequently, other mood stabilizers have been reported to affect components of this pathway.4 Abnormalities in the PI cycle are also associated with bipolar disorder.5
An important result of PI-cycle activation is an increase in intracellular calcium ion (Ca2+) signalling.6 The potential link between intracellular Ca2+ signalling and bipolar disorder is a subject of increasing interest because of growing evidence of alterations in peripheral blood cell calcium ion concentration ([Ca2+]) in patients with bipolar disorder. Elevated basal [Ca2+] has been found in platelets from unmedicated bipolar patients during mania7 and depression,8 as well as in platelets from euthymic bipolar patients treated with lithium.9–11 Elevation of [Ca2+] in platelets and lymphocytes12,13 and in B-lymphoblasts14 has been observed in patients with bipolar disorder in various states. Enhanced intraplatelet [Ca2+] was found in both unmedicated manic patients and depressed bipolar patients in response to serotonin (5-HT)11,15–17 and thrombin.7,8,18,19 5-HT-induced, thrombin-induced and thapsigargin-induced enhancement of platelet [Ca2+] also occurred in medicated bipolar patients, regardless of their state.13,16 Further support for the role of Ca2+ in bipolar disorder stems from the fact that the anticonvulsant mood stabilizers sodium valproate20–22 and carbamazepine23–26 inhibit Ca2+ channels and as a result reduce Ca2+ currents in rat neuronal cells; furthermore, clinical studies suggest that Ca2+ channel blockers may be useful therapeutic agents for some patients with bipolar disorder.27–34
In view of these previous results, we investigated the effects of dextroamphetamine, a psychostimulant used as a model of mania because it mimics symptoms of mania in healthy subjects35,36 and of the mood stabilizers lithium chloride, sodium valproate and carbamazepine on platelet [Ca2+]. Platelets were used in this study because these cells are easily accessible and share several functional similarities with neurons, including their intracellular signalling pathways.37–41
Methods
Subjects were recruited by means of advertisements within the University of Alberta. Of the 18 subjects screened, 15 were eligible for the study. All of these 15 subjects were men, 18 to 45 years of age, with no current medical or psychiatric illness and no personal history of psychiatric or cardiovascular illness. All were medication free, did not use recreational drugs and had been nonsmokers for at least 1 year. They refrained from using salicylic acid, ibuprofen, acetaminophen and any other drugs affecting platelet aggregation for at least 2 weeks before the start of the study. All subjects underwent a full physical examination, detailed medical history and electrocardiography (ECG). Potential subjects were excluded if they had high blood pressure or abnormal ECG readings. They were required to fast from midnight the previous day and were allowed to drink only water throughout each study day. Informed consent was obtained from all subjects before their participation, and the study was approved by the local ethics committee of the University of Alberta.
A double-blind, placebo-controlled, crossover design was used. Subjects came on 2 separate days 3–5 weeks apart and were given either 25 mg dextroamphetamine orally or an equivalent volume of placebo (lactose powder). Capsules were administered in a random manner such that each subject had a 50% chance of receiving either capsule on the first study day, and therefore received the alternate capsule on the next study day.
The protocol for platelet isolation and suspension was similar to one that we have used previously.42 The first 45-mL blood sample was drawn between the hours of 7 am and 8 am, and a second sample was drawn 3.5 hours after administration of the capsule. The blood was collected with a 19-gauge butterfly needle set attached to a 60-mL polypropylene syringe containing 1.9 mL citrate buffer (3.15% sodium citrate) and was processed immediately thereafter for Ca2+ measurements. For processing, the blood was transferred into a polypropylene tube to which 0.06 μg/mL prostaglandin I2 (prostacyclin, PGI2) (Sigma Chemicals, St. Louis) was added; the tube was centrifuged at 375 g for 20 minutes at room temperature. The platelet-rich plasma (18–25 mL) was removed and placed in a second tube, to which 0.3 μg/mL PGI2 was added. This tube was then centrifuged at 2240 g for 10 minutes at room temperature. The platelet-poor plasma was removed, and the platelet pellet was resuspended in 3–5 mL HEPES (4-[2-hydroxyethyl]-1-piperazine-ethanesulfonic acid) buffer (10 mmol/L HEPES, 137 mmol/L NaCl, 1.8 mmol/L CaCl2 dihydrate, 1.0 mmol/L MgCl2, 5.5 mmol/L glucose, 2.7 mmol/L KCl, 0.4 mmol/L Na2HPO4, pH 7.3) with 1.0 mol/L TRIS (2-[hydroxymethyl] aminomethane) buffer (0.67 mmol/L Trizma base, 0.30 mmol/L TRIS HCl, pH 9.0), containing 1.9 mL citrate buffer. The citrated platelet suspensions were counted with a Coulter Counter, adjusted to a concentration of 3.5 × 108 cells/mL with citrated HEPES buffer and then incubated in the dark with 3 μmol/L fura-2 acetoxymethyl ester (AM) (Molecular Probes, Eugene, Ore.) and 30 μL PF-127 (10%) (Sigma Chemicals) per 1 μmol fura-2 AM stock solution at 37°C for 30 minutes. After incubation, 0.3 μg/mL PGI2 was added, and the suspension was centrifuged at 2240 g for 10 minutes at room temperature. The supernatant was discarded, and the pellet was washed 3 times by gently layering and removing 1 mL HEPES buffer on top of the pellet. The platelets were then resuspended in 5–7 mL of HEPES buffer, counted and adjusted to a concentration of 1.0 × 108 cells/mL. All platelet suspensions were incubated for 10 minutes at room temperature in Ca2+-containing HEPES before the addition of drugs or the beginning of measurements. Platelet suspensions obtained at baseline, before the administration of any capsules, were divided into 2-mL aliquots and incubated in the dark with either 1.0 μmol/L lithium chloride (Sigma Chemicals), 600 μmol/L sodium valproate (Sigma Chemicals), 40 μmol/L carbamazepine (Sigma Chemicals), 50 ng/mL dextroamphetamine sulfate or no drug (to serve as the negative control) for 40 minutes at room temperature.
Fluorometry
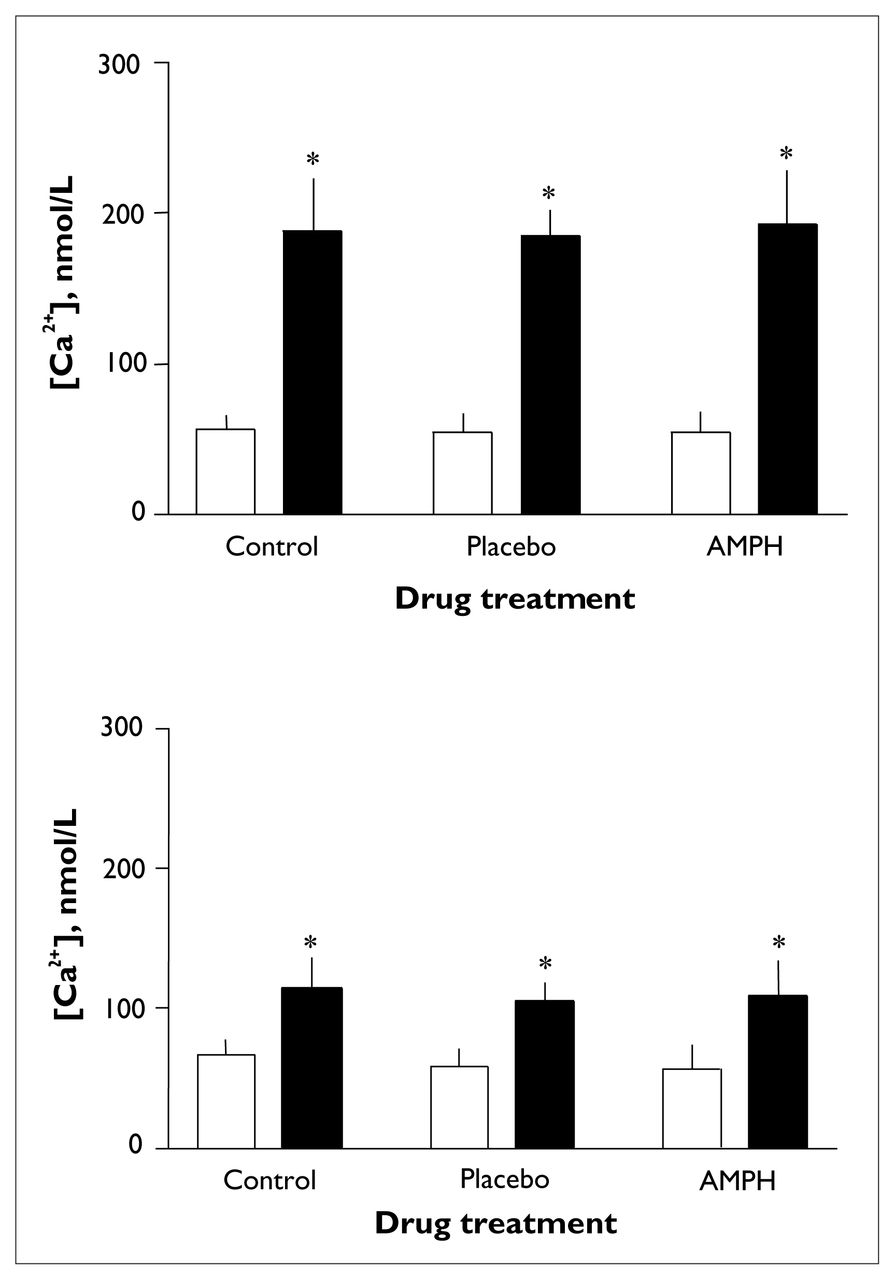
An 8100 series spectrofluorometer was used for all measurements. Samples were prewarmed to 37°C for 4 minutes in quartz cuvettes and subjected to constant gentle stirring; all measurements were taken at 37°C (in a water bath that allowed circulation of water through the sample holder). The measurements were taken within 90 minutes of the last resuspension, in duplicate, with alternation between drug-treated and untreated samples to control for any effects of time elapsed between the scans. Fluorescence intensity was recorded as a function of time, with dual excitation at 340 nm and 380 nm (the wavelengths that correspond to fura-2 bound to Ca2+ and free fura-2, respectively) and sensitivity set at 750 V. The emission wavelength was set to 510 nm. After a stable basal recording had been obtained, thrombin (0.024 U/mL) or 5-HT (10 μmol/L) was added to the samples through a small injection port located directly above the sample. This allowed continuous recording of changes in [Ca2+]. The measurements were then calibrated with the detection of minimal and maximal [Ca2+] to assess free (Rmin) and saturated (Rmax) fura-2. This was done by incubating each sample successively with EGTA (ethylene glycol-bis[β-aminoethyl]-N,N,N’,N’-tetra-acetic acid; 6.25 mmol/L) and sodium dodecyl sulfate (20 μL, 10%) (Rmin) or CaCl2 (7.5 mmol/L) (Rmax). The ratios of the peaks at 340 nm and 380 nm for both thrombin and 5-HT were used to determine the [Ca2+]. A plot of [Ca2+] versus time was automatically generated by the spectrofluorometer’s intracellular probe software program for both thrombin (Fig. 1, upper panel) and 5-HT (Fig. 1, lower panel), according to the formula below:43

Agonist-induced calcium ion concentration ([Ca2+]) in platelets at baseline and in response to 0.024 U/mL thrombin (upper) or 10 μmol/L serotonin (5-HT) (lower).
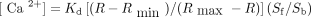
where R is the ratio of fluorescent signals at wavelengths 340 nm and 380 nm, Rmin is measured in the presence of minimal [Ca2+], Rmax is measured in the presence of maximal [Ca2+], Sf is the fluorescence of the Ca2+-free indicator measured at the 380-nm wavelength and Sb is the fluorescence of the bound indicator measured at the 340-nm wavelength. A dissociation constant (Kd) of 224 nmol/L was used,43 and Rmin and Rmax were taken directly from the time trace readings for each sample. Basal, peak and change in [Ca2+] were recorded from each graph. An excitation scan was recorded over a wavelength range of 300 nm to 420 nm at baseline and after Rmax calibration for each batch of platelets to ensure adequate dye loading. Excitation scans were automatically corrected for wavelength-dependent distortions in the intensity of excitation light and the efficiency with which it is transmitted, and 15-point smoothing was applied.
Statistical analysis
A 3-factor repeated measures analysis of variance (RMANOVA) was applied to control samples to test for differences between days, agonist effect or replicate sample. A 3-factor RM-ANOVA was subsequently used to assess the effects of treatment, agonist and replicate sample on basal and peak [Ca2+]. A 2-factor RMANOVA was applied to assess the effects of treatment and replicate sample on change in [Ca2+] (basal [Ca2+] subtracted from peak [Ca2+]). Analyses were done separately for in vivo and in vitro data, and for thrombin and 5-HT. Greenhouse–Geisser values were used to adjust the degrees of freedom for tests of significance. Tukey post hoc analysis was used for multiple comparisons (α = 0.05).
Results
There were no significant effects of day, so control data from the same subjects on day 1 and day 2 were averaged. Replicate samples were averaged because no differences between replicate samples were found.
Effects of in vivo dextroamphetamine on intraplatelet [Ca2+]
Thrombin induced a significant increase in intraplatelet [Ca2+] (F1.00 = 128.04, p < 0.001) (Fig. 2, upper panel). There were no significant differences between treatment groups (with or without dextroamphetamine) or replicate samples in terms of basal or thrombin-induced [Ca2+] or in terms of change in intraplatelet [Ca2+]. Mean basal [Ca2+] (and standard error of the mean [SEM]) were 57.3 (SEM 2.4) nmol/L for baseline controls, 54.8 (SEM 5.1) nmol/L for placebo controls and 54.1 (SEM 4.2) nmol/L for dextroamphetamine-treated samples upper). Mean peak [Ca2+] was 188.0 (SEM 10.1) nmol/L for baseline controls, 185.0 (SEM 6.8) nmol/L for placebo controls and 192.3 (SEM 10.8) nmol/L for dextroamphetamine-treated samples (Fig. 2, upper panel). Mean change in [Ca2+] was 135.3 (SEM 9.3) nmol/L for baseline controls, 130.3 (SEM 7.4) nmol/L for placebo controls and 138.2 (SEM 9.4) nmol/L for dextroamphetamine-treated samples (data not shown).

Effects of in vivo administration of dextroamphetamine on changes in platelet [Ca2+] induced by 0.024 U/mL thrombin (upper panel) or 10 μmol/L 5-HT (lower panel). Measurements at time zero (control, n = 12) or 3.5 hours after administration of placebo (n = 6) or 25 mg dextroamphetamine (AMPH, n = 11). Open bars represent basal [Ca2+] and solid bars represent peak [Ca2+]. Error bars represent the standard error of the mean. Single asterisk indicates significant increase from basal to peak [Ca2+] (p < 0.01).
5-HT-induced a significant increase in intraplatelet [Ca2+] (F1.00 = 95.78, p < 0.003) (Fig. 2, lower panel). There were no significant differences between treatment groups or replicate samples in terms of basal or thrombin-induced [Ca2+] or in terms of change in intraplatelet [Ca2+]. Mean basal [Ca2+] was 66.5 (SEM 3.0) nmol/L for baseline controls, 57.9 (SEM 5.1) nmol/L for placebo controls and 56.3 (SEM 5.2) for dextroamphetamine-treated samples. Mean peak [Ca2+] was 114.4 (SEM 6.1) nmol/L for baseline controls, 105.0 (SEM 5.1) nmol/L for placebo controls and 108.7 (SEM 8.0) nmol/L for dextroamphetamine-treated samples (Fig. 2, lower panel). Mean change in [Ca2+] was 47.9 (SEM 4.7) nmol/L for baseline controls, 47.1 (SEM 4.6) nmol/L for placebo controls and 52.4 (SEM 6.1) nmol/L for dextroamphetamine-treated samples (data not shown).
Effects of in vitro dextroamphetamine, lithium chloride, sodium valproate and carbamazepine on intraplatelet [Ca2+]
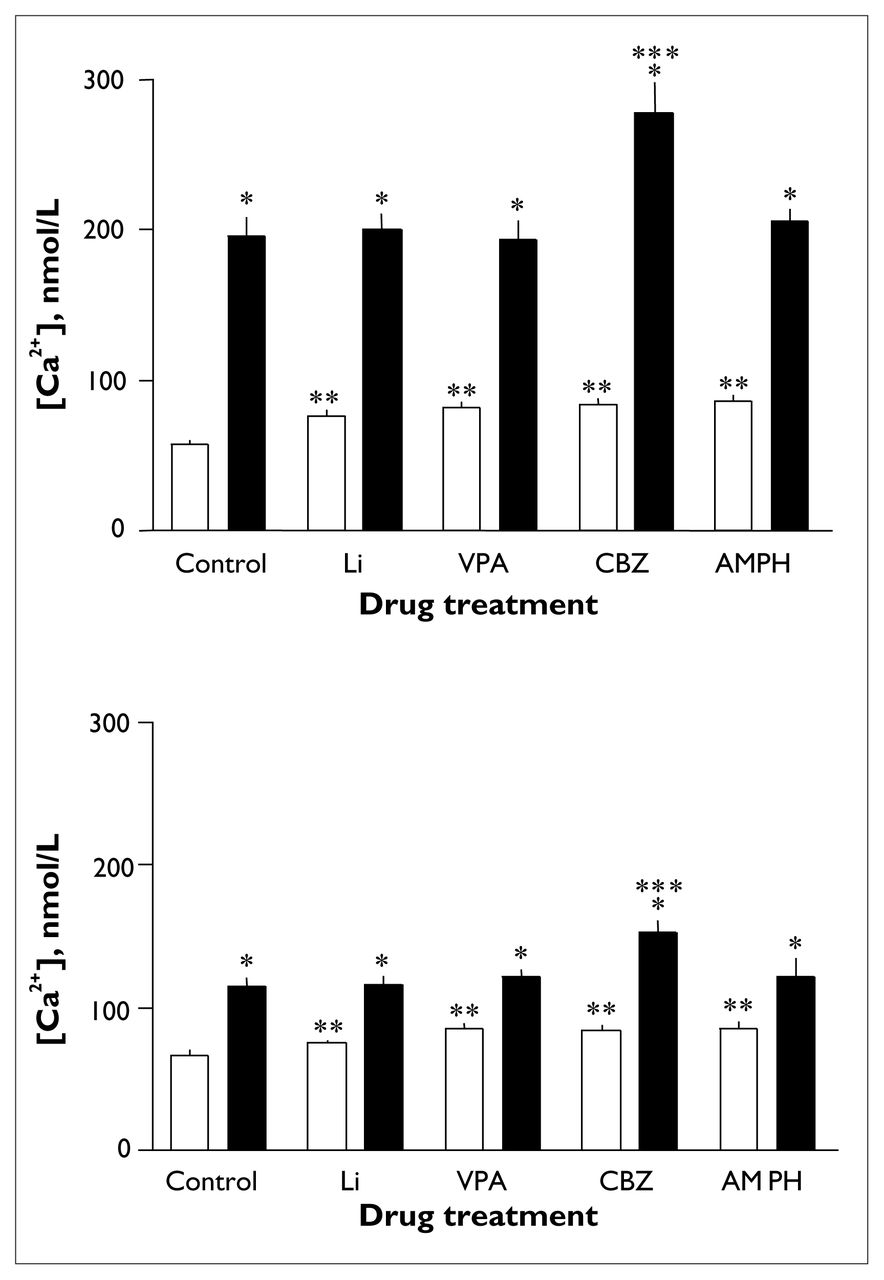
Thrombin induced a significant increase in intraplatelet [Ca2+] in all drug-treated groups (F1.00 = 170.66, p < 0.001) (Fig. 3, upper panel). In samples stimulated with thrombin, an overall effect of treatment (F2.548 = 21.76, p < 0.001) and an interaction effect of treatment by agonist (F1.772 = 42.24, p < 0.001) were found. Post hoc analyses revealed that basal [Ca2+] was significantly higher in all drug-treated groups than in untreated samples. Mean basal [Ca2+] was 58.2 (SEM 2.3) nmol/L for controls, 76.4 (SEM 3.1) nmol/L (p < 0.002) for lithium-treated samples, 82.7 (SEM 3.7) nmol/L (p < 0.001) for valproate-treated samples, 84.8 (SEM 3.3) nmol/L (p < 0.001) for carbamazepine-treated samples and 86.8 (SEM 3.9) nmol/L (p < 0.001) for dextroamphetamine-treated samples (Fig. 3, upper panel). Peak and change in [Ca2+] were similar in all groups except the carbamazepine-treated group, for which peak [Ca2+] was significantly higher than the control level (277.1 [SEM 19.9] v. 195.8 [SEM 12.2] nmol/L, p < 0.002), as was change in [Ca2+] (191.9 [SEM 19.6] v. 142.0 [SEM 10.8] nmol/L, p < 0.046) (data not shown). Mean peak [Ca2+] was 201.0 (SEM 9.4) nmol/L for lithium-treated samples, 193.1 (SEM 12.3) nmol/L for valproate-treated samples and 206.0 (SEM 8.3) nmol/L for dextroamphetamine-treated samples (Fig. 3, upper panel). Mean change in [Ca2+] was 124.3 (SEM 10.1) nmol/L for lithium-treated samples, 110.5 (SEM 10.5) nmol/L for valproate-treated samples and 119.0 (SEM 7.7) nmol/L for dextroamphetamine-treated samples (data not shown). Thrombin-induced [Ca2+] values were also significantly higher in the carbamazepine-treated group than in the lithium-treated (p < 0.004), valproate-treated (p < 0.002) and dextroamphetamine-treated (p < 0.016) groups. Change in [Ca2+] induced by thrombin was also significantly higher in the carbamazepine-treated group than in the lithium-treated (p < 0.007), valproate-treated (p < 0.001) and dextroamphetamine-treated (p < 0.008) groups (data not shown).

Effects of in vitro administration of lithium chloride, sodium valproate, carbamazepine or dextroamphetamine on changes in platelet [Ca2+] induced by 0.024 U/mL thrombin (upper panel) or 10 μmol/L 5-HT (lower panel). Measurements were made after incubation for 40 minutes at room temperature with no drug (negative control [C], n = 13), 1.0 μmol/L lithium chloride (Li, n = 10), 600 μmol/L sodium valproate (VPA, n = 11), 40 μmol/L carbamazepine (CBZ, n = 11) or 50 ng/mL dextroamphetamine (AMPH, n = 7). Open bars represent basal [Ca2+] and solid bars represent peak [Ca2+]. Error bars represent the standard error of the mean. Single asterisk indicates significant increase from basal to peak [Ca2+] (p < 0.01), double asterisk indicates significant increase in basal [Ca2+] (p < 0.01) and triple asterisk indicates significant increase in peak [Ca2+] (p < 0.01).
5-HT induced a significant increase in platelet [Ca2+] in all groups (F1.00 = 38.68, p < 0.004) (Fig. 3, lower panel). In samples stimulated with 5-HT, a significant overall effect of treatment (F2.092 = 26.34, p < 0.001) and an interaction effect of treatment by agonist (F2.552 = 33.64, p < 0.001) were observed. Subsequent post hoc analyses revealed that basal [Ca2+] was higher with sodium valproate, carbamazepine and dextroamphetamine treatment. Mean basal [Ca2+] was 67.0 (SEM 2.8) nmol/L for controls, 75.0 (SEM 2.4) nmol/L for lithium-treated samples, 85.0 (SEM 3.5) nmol/L (p < 0.002) for valproate-treated samples, 84.7 (SEM 2.9) nmol/L (p < 0.002) for carbamazepine-treated samples and 85.9 (SEM 4.3) nmol/L (p < 0.003) for dextroamphetamine-treated samples. Peak [Ca2+] and change in [Ca2+] induced by 5-HT were similar in all groups except the carbamazepine-treated group, in which the peak was significantly greater than control (153.0 [SEM 8.2] v. 115.4 [SEM 5.7] nmol/L, p < 0.003); there was also a trend toward significant difference for change in [Ca2+] (68.3 [SEM 6.0] v. 48.4 [SEM 4.3] nmol/L, p < 0.054) (data not shown). Mean peak [Ca2+] was 116.8 (SEM 5.3) nmol/L for lithium-treated samples, 121.5 (SEM 5.2) nmol/L for valproate-treated samples and 122.2 (SEM 12.2) nmol/L for dextroamphetamine-treated samples (Fig. 3, lower panel). Mean change in [Ca2+] was 41.9 (SEM 5.1) nmol/L for lithium-treated samples, 36.5 (SEM 4.1) nmol/L for valproate-treated samples and 36.3 (SEM 8.5) nmol/L for dextroamphetamine-treated samples (data not shown). 5-HT-induced peak [Ca2+] was also significantly higher in the carbamazepine group than the lithium-treated (p < 0.007) and valproate-treated (p < 0.018) groups; the difference between the carbamazepine-treated and dextroamphetamine-treated groups was nearly significant (p = 0.054). Change in [Ca2+] induced by 5-HT was also significantly higher in the carbamazepine-treated group than the lithium-treated (p < 0.01), valproate-treated (p < 0.002) and dextroamphetamine-treated (p < 0.004) groups (data not shown).
Discussion
Effects of dextroamphetamine on intraplatelet [Ca2+]
To our knowledge, this is the first study to investigate the effects of dextroamphetamine on intraplatelet [Ca2+] in human subjects. Dextroamphetamine is used as a model of mania in the study of bipolar disorder because it induces a manic-like syndrome with symptoms of euphoria, racing thoughts, increased talkativeness, goal-directed activities, self-esteem, distractibility and decreased need for sleep.35,36 Dextroamphetamine has also been associated with biological alterations that occur in conjunction with mania such as increases in heart rate and blood pressure,36 catecholamine activity, 44 cerebral metabolism,45 phosphomonoesters (a measure of inositol phosphates)46,47 and myo-inositol.47 The major action of dextroamphetamine in the brain is inhibition of the reuptake of dopamine and norepinephrine into presynaptic neurons; in addition, it increases the postsynaptic release of dopamine and norepinephrine and binds directly to postsynaptic receptors.48,49 These effects all increase the synaptic activity of the neurotransmitters dopamine and norepinephrine, both of which activate receptors that are linked to the PI cycle and hence to Ca2+ signalling.50
Since the activation of the PI cycle in neurons has many parallels to the activation of the PI cycle in platelets, 41,51 we used platelets as a model to determine the link between Ca2+ signalling and the psychological symptoms of mania (modelled by the in vivo administration of dextroamphetamine). Although there is no evidence for any direct effects of dextroamphetamine on platelets, dextroamphetamine-induced activation of cultured neurons was found to be Ca2+ dependent.52,53 Thus, although the mechanism by which dextroamphetamine may affect platelets in vivo has not been determined, the above evidence would support suggestions that this compound may induce a change in Ca2+ signalling in platelets. The fact that increased basal [Ca2+] has been associated with mania further supports this hypothesis. 7,12 We also found an increase in basal [Ca2+] in platelets incubated with dextroamphetamine. However, we did not observe any change in agonist-induced platelet [Ca2+], a finding that has also been associated with mania in previous studies.7,11,15,18 It is possible that the concentration of dextroamphetamine used (50 ng/mL, chosen to approximate peak plasma level in healthy subjects who had received a dose of 0.25 mg/kg of dextroamphetamine, which was similar to the dose we administered in this study and which thus mimicked the in vivo situation54) was not sufficient to evoke a significant change in agonist-induced platelet [Ca2+]. Alternatively, a longer incubation time might have been needed to elicit an observable response. Even though our findings are intriguing, neither basal nor agonist-induced increases in [Ca2+] are specific to mania, as these findings have also been associated with euthymia9–11 and depression7,8,17,19 in bipolar patients, as well as with other nonpsychiatric disorders such as hypertension. 55 Thus, it is necessary to determine the mechanism of these changes in platelet [Ca2+] before any connection between Ca2+ signalling, the PI cycle, mania and dextroamphetamine can be postulated.
Nonetheless, there is in vivo evidence implicating the PI cycle as a target of dextroamphetamine in the brain. One investigation showed that dextroamphetamine and lithium may target a similar pathway.46 In that study, lithium treatment for 1 week potentiated an increase in phosphomonoesters induced by dextroamphetamine in healthy subjects. Dextroamphetamine also increased myo-inositol in rat brain,56 and lithium attenuated both dextroamphetamine-induced euphoria in depressed patients57 and dextroamphetamine-induced hyperactivity in rats.58 Future studies determining the molecular targets of these behavioural paradigms would be useful.
The lack of changes in any measure of intraplatelet [Ca2+] with in vivo administration of dextroamphetamine agrees with previous studies examining the effects of dextroamphetamine on intraplatelet [Ca2+] in vivo. In a similar double-blind, placebo-controlled, crossover study in which carmoxirole, a dopamine receptor agonist, was given once daily for a week, the authors reported no changes in platelet intracellular [Ca2+] responses in healthy subjects.59 Another study reported that agonist-induced platelet [Ca2+] was not altered by smoking or physical activity (each of which is associated with increased blood pressure, heart rate, peripheral norepinephrine, epinephrine and vasopressin). 60 Finally, in vivo administration of lithium to healthy male subjects for 1, 2, 3 or 4 weeks had no effects on basal, thrombin-induced or 5-HT-induced intraplatelet [Ca2+].61 Thus, centrally acting agents may not necessarily affect peripheral blood cell [Ca2+], and, in terms of the dextroamphetamine model of mania, the short-term administration of dextroamphetamine may be insufficient to induce many of the biological changes that occur in a patient with bipolar disorder who experiences chronic manic episodes.
Effects of lithium chloride, sodium valproate and carbamazepine on intraplatelet [Ca2+]
Each mood stabilizer tested in this study increased basal intraplatelet [Ca2+] with short-term incubation at either therapeutic plasma concentrations (sodium valproate and carbamazepine) or at concentrations mimicking postulated brain levels (lithium). These findings are somewhat consistent with the inositol depletion hypothesis of lithium.3,4 Indeed, the inhibition by lithium of the activity of the catabolic enzyme responsible for the breakdown of inositol phosphates, inositol monophosphatase, causes a decrease in myo-inositol concentrations,62–68 which results in an increase in concentrations of inositol monophosphates.62–64,69–72 We postulate that this buildup of inositol monophosphates may be accompanied by an increase in inositol 1,4-triphosphate (IP3) because of the shift in the equilibrium potential that cessation of the PI cycle would cause (i.e., an increase in the products [inositol monophosphates] may lead to a decrease in the breakdown of the reactants [inositol polyphosphates]). The buildup of IP3 could cause an increase in cytosolic Ca2+ signalling via IP3-regulated Ca2+ stores. In support of this theory, both lithium67,73–77 and valproate78 increase IP3 concentrations. The acute inhibition of the PI cycle by mood stabilizers may cause a transient imbalance in the formation of second messengers. Consequently, a buildup of IP3 would signal a short-lived increase in intracellular [Ca2+]. Although these results are in agreement with our hypothesis, the PI cycle does not operate in isolation, and the numerous cellular signalling pathways (such as the cyclic adenosine monophosphate pathway) and homeostatic mechanisms (such as ion channel modulation) that would react to a significant change in PI cycle components, must be taken into consideration. Therefore, with long-term drug administration, homeostatic mechanisms may counteract this inhibition, and activation of the PI cycle may be dampened. We postulate that the changes produced by mood stabilizers when administered over the short term serve to modulate cellular events, such as transcription and translation, that would be measured differently under conditions of long-term drug treatment. Thus, we suspect that the effects seen in the acute drug-treatment paradigm are the initial events that set in motion a delayed response corresponding to the delayed therapeutic response of these drugs in the clinical setting; this suggestion is supported by reported differences in the acute and chronic effects of mood stabilizers.79–86
Our findings are inconsistent with the findings of a similar study by Kusumi et al,61 who reported that in vitro incubation of platelets with lithium, valproate and carbamazepine had no effect on basal, thrombin-induced or 5-HT-induced intraplatelet [Ca2+]; however, several methodological differences may explain this discrepancy. Of particular importance is the fact that in the earlier study61 the drugs were absent from the external media during measurements. During observation of the flux of intracellular [Ca2+] in vitro, it may be necessary for the drugs to be present in the cell suspension if they are to directly affect the pathway activated by the agonist.
Although the mechanism by which lithium inhibits the PI cycle has been well characterized,87–89 the mechanisms by which carbamazepine and sodium valproate affect this cycle are unclear. There is evidence that sodium valproate4,68,78,81,90,91 and carbamazepine92–96 affect components of the cycle. For example, sodium valproate alters Na+–Ca2+ exchange,97,98 and carbamazepine at therapeutic concentrations is a Ca2+ channel blocker.19–25 However, this is not likely the mechanism by which carbamazepine affected platelet [Ca2+] in our study, given that these voltage-gated ion channels are found in neurons but not platelets.99 The question remains as to why carbamazepine, but not lithium or sodium valproate, enhanced agonist-induced intraplatelet [Ca2+]. The answer is likely related to differences in the mechanisms of action of these 3 drugs. These drugs may act ultimately to inhibit the PI cycle, causing an acute increase in [Ca2+], but their mechanisms are probably different. The fact that we were using healthy subjects and not patients would likely play a role in determining the effects the drugs produced. In this case, the drug effect could be either to normalize platelet [Ca2+] or to make the change in platelet [Ca2+] more prominent. Future studies are needed to elucidate the precise components of the PI cycle that are targeted by these drugs and that cause changes in intracellular [Ca2+]. Furthermore, because of the differences between platelets and neurons, these studies should be replicated in neuronal cell cultures.
In conclusion, this study provides the first evidence that short-term administration of dextroamphetamine and the mood stabilizers lithium, sodium valproate and carbamazepine leads to acute increases in intraplatelet [Ca2+]. Although intracellular [Ca2+] does not appear to be involved in the in vivo dextroamphetamine model of mania used in this study, it may play a role in the therapeutic effects of mood stabilizers and the pathophysiology of mania. Further investigation of the precise mechanism by which mood stabilizers affect the PI cycle in relation to their effects on intracellular [Ca2+] may be useful in understanding the mechanisms of bipolar disorder.
Acknowledgements
This study was funded in part by grants to Peter Silverstone from the Canadian Institute for Health Research and the Alberta Heritage Foundation for Medical Research. We are grateful for the use of laboratory space and equipment provided by the Neurochemical Research Unit, the Psychopharmacology Research Unit and the Department of Pharmacology, all at the University of Alberta, and for the excellent technical assistance of Gail Rauw.
Footnotes
Medical subject headings: bipolar disorder; blood platelets; calcium; carbamazepine; dextroamphetamine; drug therapy; fura-2; lithium chloride; phosphatidylinositols; valproic acid.
Competing interests: None declared.
- Received January 29, 2002.
- Revision received July 18, 2002.
- Accepted July 29, 2002.
References

{kind=link}
{kind=link}
{kind=link}
Article tools



Related Articles
Cited By...
- No citing articles found.