

Abstract
The mechanisms by which administration of interferon-α induces neuropsychiatric side effects, such as depressive symptoms and changes in cognitive function, are not clear as yet. Direct influence on serotonergic neurotransmission may contribute to these side effects. In addition, the enzyme indoleamine 2,3-dioxygenase (IDO), which converts tryptophan into kynurenine, may play an important role, first, because IDO activation leads to reduced levels of tryptophan, the precursor of serotonin (5-HT), and thus to reduced central 5-HT synthesis. Second, kynurenine metabolites such as 3-hydroxy-kynurenine (3-OH-KYN) and quinolinic acid (QUIN) have toxic effects on brain function. 3-OH-KYN is able to produce oxidative stress by increasing the production of reactive oxygen species (ROS), and QUIN may produce overstimulation of hippocampal N-methyl-d-aspartate (NMDA) receptors, which leads to apoptosis and hippocampal atrophy. Both ROS overproduction and hippocampal atrophy caused by NMDA overstimulation have been associated with depression.
Introduction
The proinflammatory cytokine interferon-α (IFN-α) is commonly used in the treatment of patients with hepatitis C and cancer, but its administration induces neuropsychiatric side effects.1,2 Symptoms frequently associated with IFN-α treatment are fatigue, both increased sleepiness and difficulty sleeping, irritability, loss of appetite, weight loss and low mood. A full-blown depressive disorder is reported in up to 36% of cases.3 Cognitive changes involving verbal memory, cognitive speed and executive function are also reported.4–6
Administration of proinflammatory cytokines, such as interleukin-1β (IL-1β), IL-6, IFN-α and tumour necrosis factor-α (TNF-α), in rats or mice induces a behavioural pattern referred to as “sickness behaviour,” 7 which includes increased sleep,8 reduced locomotor activity, 9,10 decreased sucrose consumption (as a measure for anhedonia),11 weight loss and decreased social exploration. 12 These symptoms resemble the vegetative symptoms of depression in humans. 2,13
The development of sickness behaviour14 and depression15, 16 is linked to dysregulation of the neurotransmitter serotonin (5-HT). Several neurochemical changes in both the peripheral and central 5-HT system are seen in depressed patients, such as lower levels of peripheral tryptophan (TRP), the precursor of 5-HT;17 changes in central 5-HT transporter (5-HTT) function;18 and changes in 5-HT1A 19 and 5-HT2A brain receptors.20 Therefore, one way in which IFN-α may induce depressive symptoms is by affecting the serotonergic system.
IFN-α and central 5-HT neurotransmission
IFN-α exerts direct influence on 5-HT brain neurotransmission. IFN-α and other proinflammatory cytokines, such as IFN-γ, TNF-α and IL-1, have been shown to upregulate 5-HTT, causing a decrease of extracellular 5-HT.21–23 IFN-α may also indirectly influence 5-HTT activity by increasing the production of IFN-γ, TNF-α and IL-1. In addition, IFN-α modulates the 5- HT1A 24 and 5-HT2 25 brain receptors.
IDO activity causes TRP depletion
A second pathway by which IFN-α modulates the 5- HT system is by induction of the enzyme indoleamine 2,3-dioxygenase (IDO). IDO is the rate-limiting enzyme in the l-TRP–kynurenine pathway that converts l-TRP, the precursor of 5-HT, to N-formylkynurenine, resulting in a diminished synthesis of central 5-HT. IDO is widely distributed in various tissues, including the brain, lung, heart, kidney and intestine.26
Cytokines, such as IFN-α, IFN-β, TNF-α and IFN-γ, have been shown to upregulate IDO expression.26–31 IFN-α has a weak direct effect on IDO induction and an indirect effect through a 15-kD protein, which is a product of IFN-α-stimulated monocytes and lymphocytes and stimulates IDO and IFN-γ production.32
IFN-α therapy in patients with hepatitis C causes a decrease in TRP (4–6 months after starting therapy) and an increase in kynurenine plasma levels seen at 2 weeks after starting therapy, with kynurenine plasma levels remaining the same as at 2 weeks when measured at weeks 4, 16 and 24, indicating higher IDO activity.33 In addition, IDO induction is also mediated by an IFN-γ-independent mechanism involving the proinflammatory cytokine TNF-α 26 Anti-inflammatory cytokines, such as IL-4 and IL-10, diminish TRP metabolism.34
Overstimulation of IDO leads to depletion of plasma concentrations of TRP and, therefore, to reduced synthesis of 5-HT in the brain,35 which may play a role in the development of depressive symptoms. In addition, not only TRP but also 5-hydroxytryptophan (5-HTP) and 5-HT itself can be substrates for IDO.36 Therefore, it can be postulated that in addition to lowering peripheral levels of TRP, 5-HT synthesis in the brain can also be reduced by central degradation of 5-HT by IDO. However, there is another possible way in which IFN-α-induced overstimulation of IDO may produce depressive symptoms. By upregulating IDO expression, IFN-α can initiate the kynurenine pathway leading to the production of a variety of neuroactive metabolites. These kynurenine metabolites themselves may play a causative role, because the ratio of kynurenine to TRP is positively associated with depression and anxiety scores.33,37
Neurotoxic metabolites of the kynurenine pathway
Several metabolites formed along the kynurenine pathway are found to have neurotoxic effects, such as 3-hydroxy-kynurenine (3-OH-KYN), a direct metabolite of kynurenine, and quinolinic acid (QUIN), which is formed later in the kynurenine pathway.38–42 Peripheral kynurenine is transported through the blood–brain barrier by a large neutral amino acid carrier and thus may easily reach the central nervous system. In the brain it is taken up by glia cells, by which it is further metabolized.43,44 In this way, neurotoxic metabolites are formed in the brain that can cause neurodegeneration.
The neurotoxicity of these kynurenine metabolites has been demonstrated in animals45–50 and in vitro,41 whereas data for humans show elevated levels in several degenerative disorders. Increased production of 3- OH-KYN or QUIN, or both, is found in certain neurodegenerative conditions in humans,38 such as in Huntington’s disease,51,52 Parkinson’s disease53 and in the AIDS–dementia complex.54,55 In the last condition, QUIN levels are increased in the cerebrospinal fluid up to 20-fold and are correlated with the severity of cognitive and motor dysfunctions. Increased production of 3-OH-KYN and QUIN may also contribute to neuronal damage in cognitive decline of aging,56 infections of the central nervous system,57 malaria,58 ischemia, 59 hypoxia at birth,60 traumatic injury61 and epilepsy.62 In addition, they may play a role in the development of psychiatric diseases such as anxiety,63 depression64 and schizophrenia.65
The following mechanisms may account for the effect of 3-OH-KYN and QUIN on neurodegenerative diseases and depressive symptoms (Fig. 1). Even relatively low levels of 3-OH-KYN may cause neurotoxicity by inducing oxidative stress and neuronal apoptosis.40,41 3-OH-KYN is transferred into cells by neutral amino acid transporters. Only after interaction with cellular xanthine oxidase is 3-OH-KYN capable of producing sufficient amounts of reactive oxygen species (ROS), such as superoxide radical, hydrogen peroxide and hydroxyl radical, to induce internucleosomal DNA cleavage leading to apoptosis. Various antioxidative agents prevent cell death induced by 3-OH-KYN. The cortex and the striatum are most sensitive to 3-OH-KYN insults. Differences in vulnerability to 3-OH-KYN for different brain regions are likely to result from differences in their ability to take up large neutral amino acids.41 Overproduction of ROS has been associated with depression. An association between overproduction of ROS and increased monoamine oxidase (MAO) activity has been suggested.66 In addition, polyunsaturated fatty acids (PUFAs) are vulnerable to oxidation. Overproduction of ROS might result in destruction of phospholipids and reduce the viscosity of cell membranes. 67 Alterations in membrane viscosity may influence receptor density or function of serotonergic or catecholaminergic receptors.68 Overproduction of proinflammatory cytokines, increased MAO activity and, thus, lower levels of catecholamines, disturbances in PUFA structures and ratios, and the decrement of serotonergic and catecholaminergic receptor densities and functioning are all associated with depression.2,69,70 Furthermore, selective serotonin reuptake inhibitors have antioxidant properties and reverse the overproduction of ROS.71
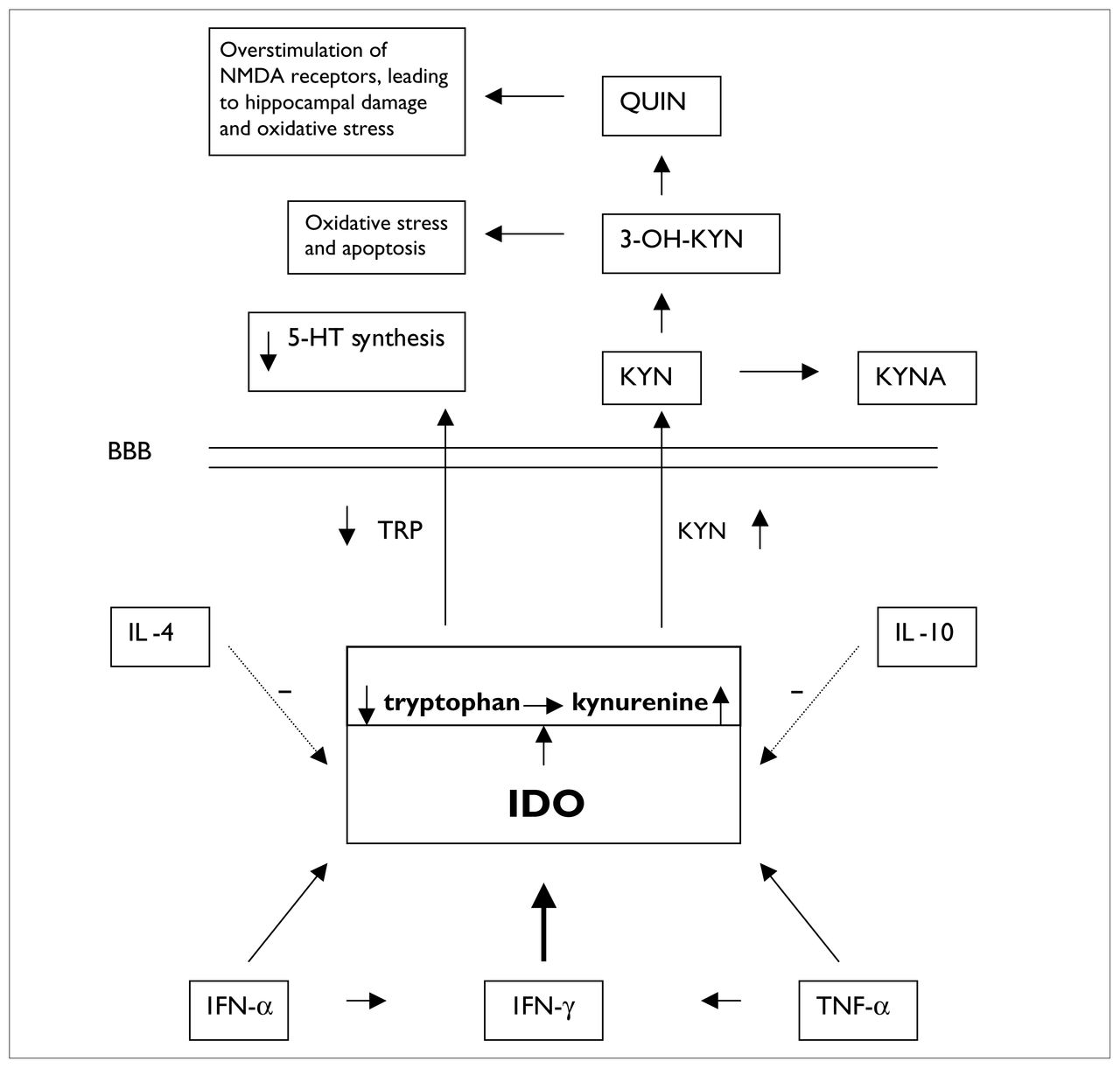
The role of indoleamine 2,3-dioxygenase (IDO) in cytokine-induced lowered tryptophan (TRP) levels and production of neurotoxic metabolites. The dotted arrows indicate that IL-4 and IL-10 diminish IDO activity. The diagonal arrows leading from IFN-α and TNF-α to IDO indicate that these cytokines increase IDO activity and, thus, cause increased metabolism of TRP, resulting in decreased availability of TRP that crosses the BBB to synthesize 5-HT. The horizontal arrows between IFN-α and IFN-γ and between TNF-α and IFN-γ refer to the fact that IFN-α and TNF-α increase production of IFN-γ, which exerts a powerful stimulating effect (thick vertical arrow) on IDO activity. NMDA = N-methyl-d-aspartate, QUIN = quinolinic acid, 3-OH-KYN =3-hydroxy-kynurenine, 5-HT = serotonin, KYN = kynurenine, KYNA = kynurenate, BBB = blood–brain barrier, IL-4 = interleukin-4, IFN-α = interferon-α, TNF-α = tumour necrosis factor-α.
QUIN is a potent N-methyl-d-aspartate (NMDA) receptor agonist. Overstimulation of NMDA receptors increases calcium influx into the target neurons, which will lead to neuronal damage.47,72 In addition, QUIN can contribute to the formation of free radicals, first, because the influx of calcium ions into neurons after activation of glutamate receptors may lead to the generation of ROS.73,74 Furthermore, with the generation of free radicals, QUIN induces lipid peroxidation in membrane lipids and proteins, leading to changes in neuronal membrane fluidity, receptor function and ion permeability.75 Finally, QUIN and 3-OH-KYN act synergistically in the production of free radicals, so that even low doses of QUIN potentiate the excitotoxicity of 3-OH-KYN.76,77
Intrahippocampal injection of QUIN in rats causes substantial loss of hippocampal neurons.45,48,75 QUIN-induced neurotoxicity can be significantly reduced by pharmacologic blockade of kynurenine 3-hydroxylase, the enzyme responsible for the formation of 3-OH-KYN from l-kynurenine or by administration of kynurenate (KYNA), another metabolite of the kynurenine pathway, which inhibits NMDA receptor function and thus protects against excitotoxic insults.48
Hippocampal neurodegeneration and depression
Major depression is associated with hippocampal volume loss.78,79 Hypothalamic-pituitary-adrenal (HPA) axis inhibition appears to be mediated by negative feedback from circulating glucocorticoids.80 Destruction of the hippocampus attenuates the negative feedback via loss of glucocorticoid receptors, which will increase HPA activity. Loss of the normal glucocorticoid feedback has been reported in humans with depression and animals in chronic stress paradigms.81–83 Hippocampal atrophy results from an excess of excitatory amino acid neurotransmitters, such as glutamate that acts on the NMDA receptor, which can be produced by glucocorticoid overexposure.84–86 However, Magariños and McEwen87 found a dissociation between parameters indicating the glucocorticoid stress response and hippocampal atrophy in rats. Dendritic atrophy occurred in spite of the habituation of the glucocorticoid stress response after days of repeated stress. In addition, hypercortisolism occurs in about half of the patients with depression, whereas atrophy seems to be demonstrable in a far higher percentage of individuals.86 This suggests that, besides glucocorticoids, other factors contribute to neurodegeneration, for example, the neurotoxic metabolites of kynurenine, whose levels are raised in depression. Therefore, we assume as a hypothesis a second mechanism in which kynurenine metabolites, such as QUIN, cause NMDA receptor overstimulation, leading to hippocampal atrophy and subsequent interference with the normal negative feedback function of HPA axis activity.
Conclusion
Administration of IFN-α causes the development of depressive symptoms in a high percentage of patients. This may be caused by direct influence of IFN-α on central 5-HT transmission, or indirectly via induction of the enzyme IDO. The latter may play an important role in the pathophysiology of IFN-α-induced depression through its effect on brain TRP availability that is crucial to the formation of central 5-HT. Furthermore, IDO is the rate-limiting step in the brain kynurenine pathway that leads to the formation of neurotoxic substances, such as 3-OH-KYN and QUIN, which cause neurodegeneration that may contribute to the development of depression.
Footnotes
Medical subject headings: depression; interferon-alpha; serotonin; tryptophan.
Competing interests: None declared.
- Received October 29, 2002.
- Revision received March 6, 2003.
- Accepted March 17, 2003.
References

{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.