

Abstract
The accessibility of cognate binding sites within a gene promoter can be modified by the condensation or relaxation of local chromatin structure. Local chromatin structure is in turn programmed by covalent modifications of cytosine bases in DNA and amino acid residues in histone protein tails. These chemical and physical adaptations around gene promoters can significantly change levels of mRNA expression. Furthermore, linear patterns of covalent modification of histone protein tails are emerging as a distinct regulatory code — another form of cellular memory. Because chromatin structure can be modified by conventional pharmacologic therapy, a novel approach to the regulation of neuronal gene expression in clinical populations is possible.
Introduction
Chromatin, a DNA–protein complex, while representing the efficient “packaging” of several billion bases of genomic DNA, can also function as an interactive platform for the regulation of gene transcription. Chromatin participation in gene regulation is based on physical and chemical adaptations in the vicinity of regulatory DNA sequences, the mechanics of which are determined by linear patterns of covalent modifications of cytosine bases in DNA and amino acid residues in histone protein tails. These covalent modifications, along with their attendant enzymes and cognate regulatory proteins, are broadly classified under the general term “epigenetic mechanisms.”
Historically, epigenetic mechanisms were considered relevant to the dividing cell, both in normal development and in cancer. Variation in CpG (cytosine–phosphodiester–guanine) methylation as a basis for mental disorders is a relatively new proposition.1,2 A series of discoveries has now thrust these mechanisms squarely in the domain of research on schizophrenia, bipolar disorders and other neuropsychiatric illnesses. As a first step, several neurodevelopmental syndromes have been incontrovertibly ”linked” to pathological modifications of epigenetic events, e.g., Rett syndrome, with multiple mutations in the canonical methylated CpG (5mCpG) binding protein MeCP2,3 and fragile X syndrome, caused by expanded CGG repeats in the fragile X gene promoter, which are susceptible to CpG methylation and subsequent promoter downregulation.4 Second, there has been the surprising observation that postmitotic neurons (but not mitotic oligodendrocytes or astrocytes) are especially rich in the prototypic DNA methylating enzyme DNMT1 (DNA methyltransferase 1).5,6 DNA methylation is now considered vital for successful brain development and neuronal maturation. 7 Third, it has recently been discovered that valproic acid, the drug most widely prescribed to treat bipolar disorder, is an inhibitor of histone deacetylases (HDACs) when used in clinically relevant concentrations.8 HDACs are key enzymes in the modification of chromatin structure. Fourth, we have observed in our laboratory that the regulation of candidate genes relevant to psychiatric disorders, such as reelin (RELN) and GAD67, is explicitly influenced by variations in promoter methylation and histone modification.9–12 Finally and most recently are the reports that the expression of DNMT1 is significantly increased in cortical γ-aminobutyric acid (GABA)-ergic interneurons taken post mortem from brains of psychotic patients with schizophrenia and bipolar disorder.6,13
DNA methylation in neurons
DNA methylation in the brain and other tissues is mediated by the catalytic activities of DNA methyltransferase enzymes, including DNMT1, DNMT3a and DNMT3b. 5mCpG in turn serves as an attachment site for members of the family of methylated CpG binding domain (MBD) proteins, of which MeCP2 is an archetype.
DNMT1
The level of DNA methylation in humans is higher in the brain (0.98 mole percent of 5mCpG) than in the lymphocytes, liver, spleen, lungs, liver, heart, sperm or placenta (in declining order;14 see Tawa et al15 for evidence of a similar pattern in mice). Around the time of birth, there are significant changes in the patterns of 5mCpG markings, the markings generally decreasing with differentiation.16 Treatment of undifferentiated progenitor cells with demethylating agents such as 5-azadeoxycytidine17 or retinoic acid (which affects chromatin structure) induces specific differentiation characteristics for cholinergic, dopaminergic and noradrenergic neurons. Whereas the DNMT1-null mouse is embryonic lethal, the conditional deletion of the DNMT1 gene using the cre-lox recombinase system reveals important aspects of the neuronal function of this protein.7 If gene deletion is engineered in postmitotic cerebellar granule cell cultures, DNMT1 protein levels are undetectable by day 3, which indicates rapid protein turnover. When gene deletion is engineered in vivo in neuronal precursor cells (at day E9 to E10), then deletion-containing neurons are able to survive through embryogenesis. However, these neurons are hypomethylated, and mutant embryos carrying 95% hypomethylated neurons die at birth because of respiratory failure.7 If the cre-lox recombinase method is modified to generate mutant embryos with only 30% hypomethylated neurons (known as “mosaic brain”), the organism typically survives, but the hypomethylated neurons are selectively eliminated in the early postnatal weeks.7 Finally, when the DNMT1 gene is deleted from differentiated postmitotic neurons of transgenic mice during the perinatal stage, DNMT1-deficient neurons are observed to survive for up to 17 postnatal months.7 Together, the findings from this comprehensive study suggest that DNMT1 is responsible for genomic methylation patterns in neuronal precursors, and that disruption of these patterns gives rise to neurons that do not survive. However, it also seems evident that after neuronal differentiation has occurred, DNMT1 is no longer necessary to maintain global DNA methylation, and abrogation of DNMT1 activity fails to influence long-term survival. Nonetheless, and especially given that DNMT1 is abundantly expressed in adult differentiated neurons, particularly GABAergic interneurons,6 the role of DNMT1 in the understanding of gene regulation at the level of single gene promoters or even of a single 5mCpG site needs further investigation. We have now demonstrated in primary cortical cultures (83% expressing GABAergic markers) that a decrease in DNMT1 mRNA and protein induced by DNMT1 antisense oligonucleotides results in upregulation of RELN promoter activity.12 Surprisingly, mice with reduced levels of neuronal DNMT1 showed less neuronal damage after vascular injury than wild-type mice, which suggests that enhanced gene expression due to reduced DNA methylation may facilitate neuronal survival.18
Understanding the “pattern” of 5mCpG markings that is most predictive of transcriptional activity has proved difficult. The question here is whether the coarse density at a given locus (assayed with methylation-sensitive restriction enzymes) is more informative than a linear pattern of site-specific markings analyzed at the single-nucleotide level (assayed using bisulfite modification). There is also the question of which locus should be given priority: a locus within the sequence of a transcription factor binding site or one elsewhere along the gene, such as in the first exon or perhaps even the occasional intron. Walsh and Bestor,19 using methylation-sensitive restriction enzymes to compare 5mCpG densities on genes from expressing and non-expressing tissue, provide numerous examples in which gross DNA methylation patterns were not predictive of gene expression. The bisulfite-modified DNA sequencing method can capture a 5mCpG marking pattern at the single-nucleotide level and may provide answers to these questions. Nonetheless, there is one key criterion that has not yet been established, and that is whether the imposition of a methylating pattern from a nonexpressing tissue can shut down transcription in a fully expressing tissue.19 A second criterion that has proved elusive is the identification of a protein endowed with demethylase activity, a component that would be critical for dynamic regulation; this is considered in a later section of the current review.
MeCP2
The 5mCpG sites within gene promoters are targeted by MBD-containing proteins such as MeCP2, a protein that is also expressed more abundantly in brain than in other tissues. Interestingly, MeCP2 is preferentially expressed in neurons rather than glia. Within neurons, MeCP2 is prominently expressed in the nucleus but can also be identified in postsynaptic cytoplasm, a characteristic that is shared with other transcriptional regulators such as c-fos and c-jun.20 Localization of a transcription regulator (mRNA or protein) in the postsynaptic cytoplasm strongly suggests recruitment of this regulatory protein by synaptic activity followed by translocation to the nucleus to regulate gene expression. Within the nucleus, promoter-bound MeCP2 recruits corepressor complexes containing histone deacetylases and chromatin-remodelling proteins (Fig. 1). The outcome is a local condensation of chromatin around the targeted gene promoter, which results in transcriptional repression. MeCP2-induced chromatin condensation appears to be a dynamic process because neuronal depolarization causes a release of MeCP2 from its binding site and consequently a decondensation of chromatin around that particular site21,22 (see Fig. 1). Functional mutations in the MeCP2 gene are almost exclusively expressed as neuronal phenotypes; this is inferred from human subjects affected by Rett syndrome or from MeCP2-null mice. Because vulnerability specific to neuron tissue is not accompanied by major changes in neuron gene expression, the role of neuronal MeCP2 is not immediately evident from current studies of its 5mCpG-binding properties. One possibility includes a unique role for MeCP2 in the expression and control of neuronally restricted gene sets.23 Alternatively, MeCP2 may be a non-unique contributor to the stemming of transcriptional “leakage” in neurons, a prerequisite for the high-fidelity processing of multiple synaptic inputs. Because transcription profiling of MeCP2-mutant neurons demonstrates only subtle changes in levels of gene expression, a role in noise reduction is increasingly supported.24
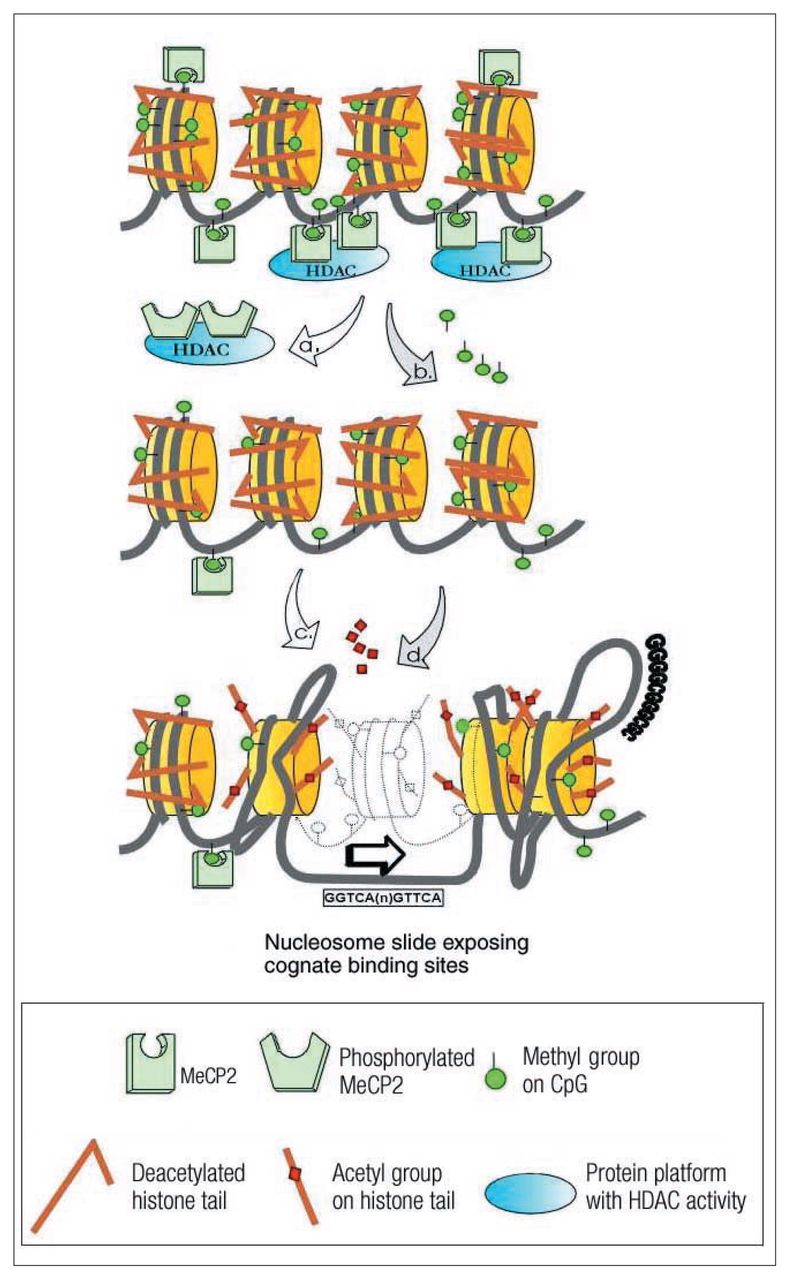
Top: Chromatin structure in quiescent nondepolarized neurons. MeCP2 is attached to methylated CpG sites and recruits protein complexes containing histone deacetylases (HADCs). The HDACs maintain the histone protein tails in a deacetylated state, and as a consequence the chromatin structure remains condensed. Middle: Depolarization induces 2 processes: phosphorylation of MeCP2, which changes the affinity for methylated CpG sites and results in disengagement of MeCP2 and HDAC-containing protein platforms (indicated by “a”); and demethylation of specific CpG sites (indicated by “b”). Bottom: Covalent modification of histone protein tails occurs by acetylation of specific amino acid residues, which results in disengagement from nucleosomally wrapped DNA (indicated by “c”). In addition, nucleosome remodelling proteins induce “sliding” of the nucleosome assembly and “buckling” of DNA strands, both of which result in exposure of cognate binding sites (e.g., nuclear receptors, SP1 binding sites) and transcription initiation sites (indicated by “d”).]
Molecular links between DNA methylation and chromatin remodelling
Activation or repression of gene transcription is correlated with the acetylation status of nucleosomal histone proteins, especially those in the vicinity of gene promoters. The enzymes that perform histone acetylation (histone acetyltransferase, HAT) or histone deacetylation (HDACs) are usually associated with large multiprotein complexes. The binding of MeCP2 to methylated DNA sites in gene promoters initiates a series of events, including recruitment of HDACs, deacetylation of histone protein tails and chromatin condensation around the gene promoter, causing transcriptional repression (Fig. 1).
Three classes of proteins with HDAC activity (classes I, II and III) comprising a total of 18 members25–27 have been identified. The class I HDAC enzymes are nuclear enzymes commonly associated with protein platforms such as NuRD (nucleosome remodelling and histone deacetylation) and Sin3.28 The physiologic regulation of HDAC activity depends on phosphorylation, corepressor activity and shuttling of enzyme between cytoplasm and nucleus.26 Although whole brain tissue expresses HDACs 1, 3, 4, 5, 6, 7, 9 and 10, regional and cellular differences in the brain expression of specific members of this family can be expected. Hence, cell-specific enrichment of a particular HDAC enzyme subtype coupled with an increasing array of HDAC inhibitors (of which valproic acid is a clinical prototype) suggests the therapeutic possibility of inhibiting HDAC activity in a defined cell type localized to a particular brain area.11
Covalent modifications of N-terminal histone protein tails are not limited to acetylation, but can include methylation, phosphorylation and ubiquitination. Amino acid residues along histone protein tails emerging from the octomeric nucleosome assembly, especially arginine and lysine, are subject to site-specific acetylation and methylation. Specific linear combinations of acetylated and methylated sites are associated with “open” or “closed” chromatin formations and are now termed the “histone code.” This “code” mediates protein–protein interactions contributing to the short-term and long-term regulation of transcription and, furthermore, may represent the coding for a specific form of cellular memory. For example, dimethylation of lysine in position 9 of the H3 histone protein tail (H3dmK9) is associated with the formation of closed chromatin, whereas acetylation at the same site (H3aceK9) or dimethylation of lysine in position 4 (H3dmK4) is associated with “open” or relaxed chromatin.
Thus, the acetylation of histone protein tails can locally expand chromatin in the region of specific promoters without disrupting the structure of the core particle of the nucleosome. Binding of regulatory transcription factors is facilitated by displacement of the DNA strand from intact core particles29 (Fig. 1). Indeed, both promoters and enhancers of active genes are usually seen in DNA regions dissociated from nucleosomes. Nucleosomes are displaced from these regions by chromatin remodelling “machines” in 2 representative ways. The SWI/SNF (switch/sniff remodelling proteins) complex disrupts DNA histone contacts by inducing torsion and buckling of the DNA strand,30 whereas the NURF (nucleosome remodelling factor) family of nucleosome modellers “slides” the histone away from regulatory sites31 (Fig. 1).
The possibility of a “targeted” approach to chromatin remodelling is also increased by the existence of ligand-activated nuclear receptors (LNRs), which recognize specific DNA sequences in gene promoters. These receptors may indeed function as “epigenetic switches,” inducing expression in the liganded state and inhibiting expression in the ligandfree state by changing local chromatin structure.32 Promoter-bound LNRs will recruit coactivators (such as nuclear receptor coactivator [N-CoA] and steroid receptor coactivator [SRC-1]), which in turn recruit multiprotein complexes containing HAT molecules such as P300/CBP/PCAF. Finally, HATs acetylate histone protein tails and induce a relaxed chromatin structure conducive to transcription.
Depolarization-induced changes in DNA methylation and chromatin structure
Activity-dependent (or depolarization-induced) gene regulation is the sine qua non of neuronal function, forming the basis by which the central nervous system captures transient experiential stimuli and transforms them into long-term biochemical changes. A key question now under investigation is whether epigenetic mechanisms participate in depolarization-induced changes in gene regulation. The gene for brain-derived neurotrophic factor (BDNF) has been well studied; its transcription is dramatically upregulated by membrane depolarization, and its protein product effects changes in neuronal survival and plasticity. It is now reported that depolarization induces BDNF transcription through a series of epigenetic modifications (Fig. 1). Chen et al21 demonstrate that in the quiescent neuron, MeCP2 binds to the BDNF promoter and acts as a negative regulator of BDNF expression. In response to membrane depolarization-induced calcium influx through L-type voltage-sensitive calcium channels, MeCP2 becomes phosphorylated, a process that reduces the affinity of this protein for 5mCpG sites in the promoter. Because MeCP2 mediates long-term gene silencing through changes in chromatin structure, the release of MeCP2 from the BDNF promoter results in predictable modifications in chromatin structure. In a quiescent neuron, chromatin around the BDNF promoter is rich in histone H3 dimethylated at Lys9 (H3dmK9), which is strongly indicative of a repressed transcriptional state. Membrane depolarization causes a reduction in dimethylated H3 (H3dmK9) and an equivalent increase in acetylated H3 at Lys9 (H3aceK9), which represents a transition from a repressive to a permissive chromatin structure. In a conceptually related study, Martinowich et al22 report that site-specific methylation of CpG dinucleotides in the BDNF promoter is associated with reduced expression and that a lower level of promoter methylation is associated with upregulation of BDNF expression. Membrane depolarization induces a dissociation of MeCP2 from the BDNF promoter in a gene-specific manner, i.e., MeCP2 does not dissociate from the promoter of an imprinted control gene. Membrane depolarization again resulted in demethylation of the BDNF promoter and in comparable changes in histone biochemistry (from histone H3 dimethylated at Lys9 [H3dmK9] to H3 acetylated at Lys14 [H3aceK4]).22 These studies demonstrate depolarization-induced changes in DNA methylation and chromatin structure, as well as the modification of epigenetic regulatory proteins such as MeCP2, in a time frame of several hours. The effects of membrane depolarization on histone acetylation status are particularly noteworthy because Huang et al33 have shown that experimentally induced seizures result in altered histone acetylation in the vicinity of the promoters of the BDNF and glutamate receptor 2 (GluR2) genes.
Demethylase activity
A much-debated issue is whether CpG methylation acts as a primary transcriptional regulator or if it provides a quasi-stable “genomic floor plan” specific to the tissue type (e.g., neurons or hepatocytes) by partitioning genes into sets that are never, sometimes or always transcribed. In dividing cells, 5mCpG markings are somatically (mitotic) and, in specific instances, genetically (meiotic) stable. In postmitotic neurons, genes that have been silenced by imprinting, by inactivation of the X chromosome or by repression of viral genomes (situations that use CpG methylation) are not likely to be reactivated, which suggests structural rather than dynamic control. The debate essentially hinges on the demethylating properties of the cell and whether the known mechanisms are adequate for rapid demethylation and erasure of the 5mCpG mark.34 The most widely accepted demethylating mechanism in postmitotic cells is the excision of the methylated cytosine base and repair of the DNA strand, a process generally considered too cumbersome for facile regulation. Consequently, there has been a concerted effort to identify a demethylating protein that can remove the methyl group directly from the cytosine ring, which would obviate the need for DNA resynthesis. In a sequential series of published experiments, just such a protein was initially identified from a cDNA library and was subsequently found to code for MBD2b, a previously known transcriptional repressor.35,36 However, work based on these observations has run aground, primarily because other groups have not been able replicate them,37–39 but also because the enzymatic reaction proposed is questionable on the basis of modern chemical reaction theory.34 At this juncture, there is no consensus on the actual sequence identity of a putative demethylase. However, the reports reviewed above suggest that demethylation of particular 5mCpG sites on promoters is observable in studies of neuronal activity after stimuli of a few hours to days. The mechanism of this “active” demethylation is as yet undetermined.
Gene expression: DNA methylation, histone deacetylation or chromatin remodelling?
A synergy among the intensity of DNA methylation, the extent of histone protein tail acetylation and the activity of chromatin remodelling complexes appears to depend on individual promoters. Cameron et al40 have demonstrated that in transformed cell lines, the stable maintenance of a silent state depends primarily on DNA methylation, i.e., intense CpG methylation of the promoter strongly predicts a transcriptionally inactive gene that would be resistant to any covalent changes of the histone protein tails. However, at a modestly lower level of methylation, promoter activity can be promulgated by reducing the acetylation of histone protein tails through administration of HDAC inhibitors such as trichostatin-A. The next level of promoter regulation is the interplay between acetylation status and recruitment of remodelling complexes, which in turn makes available various cis-acting regulatory elements that are recognized by sequence-specific transcription factors. Chromatin remodelling and histone acetylation are complementary processes, and individual promoters may dictate a particular sequence of events (for a review, see Neely and Workman41). For example, at the human interferon-β and estrogen-receptor-dependent genes, histone acetylation precedes chromatin remodelling. Conversely, the prior recruitment of chromatin remodelling proteins followed by recruitment of HATs is a sequence that is readily observed with promoters encapsulated in dense chromatin, such as in mitotic cells.41 Ultimately, these cooperative mechanisms create a local chromatin structure that is readily accessible to the assembly of transcription factors required to recruit the RNA polymerase machinery and initiate transcription.
Encoding of experience and long-term memory by epigenetic events
From a neuroscience perspective, the necessary question is whether the epigenetic events described above actually participate in aspects of brain function that are relevant to behavioural scientists and clinicians. We consider here the observation from a recent paper that puts into context the translation of complex maternal behaviour and early developmental experiences into long-term epigenetic modification along the promoter of genes expressed in the neonatal hippocampus, resulting in long-term changes in neuronal gene expression. Weaver et al42 demonstrate that the style of parenting of neonatal rat pups by mothers who engage in intensive licking and grooming behaviours results in DNA demethylation and histone covalent modification along key regulatory sites within the promoter DNA sequence of a glucocorticoid receptor gene in the offspring.
More direct evidence substantiating the role of chromatin remodelling in the maintenance of long-term memory and synaptic plasticity is provided by 2 reports on a mouse model of the Rubinstein–Taybi syndrome, which is characterized by mutations in the cAMP-responsive element binding protein (CREB) binding protein (CBP). The CBP possesses properties of a HAT and can therefore remodel chromatin. Alarcon et al43 report that cbp+/− heterozygous mutants are deficient in levels of acetylated histone 2B, late long-term potentiation, and long-term memory for fear and object recognition. These deficiencies are reversed with the use of an HDAC inhibitor. Korzus et al44 demonstrate the effects of chromatin remodelling in the stabilization of long-term memory using transgenic mice expressing CBP with the HAT property eliminated but the protein–protein interaction domains or platform function retained. Activation of the transgene results in impairment of long-term memory that is reversible with the administration of an HDAC inhibitor.
Pharmacologic approaches to chromatin remodelling
The identification of HDAC inhibitors, such as valproic acid (as in Depakote), heralds a new era of therapeutics, specifically, conventional pharmaceuticals that directly target genomic structures such as chromatin. A treatment strategy for schizophrenia based on epigenetic mechanisms is now firmly supported by evidence at the molecular level,2,45 and the beneficial effects of valproic acid in this illness have been documented. 46 An immediately applicable strategy is the use of valproic acid as a genome “softener,” especially in treatment-resistant cases where the usual treatment algorithms, including last-ditch combinations of multiple drugs from different classifications, have failed. The increased plasticity of the genome could reinvigorate treatment response in these refractory cases. Indeed, it is tempting to speculate that these and similar agents may enhance the effects of psychiatric rehabilitation programs and cognitive retraining in patients with long-term disabilities.
Early pharmacologic efforts at demethylating genomic DNA involved use of nucleoside analogues (e.g., 5-azadeoxycytidine). The inherent difficulty with these compounds is the problem of dynamic remethylation of genes that were demethylated with these agents. Consequently, the identification of HDAC inhibitors able to target a tissue-specific HDAC enzyme is a significant priority. Among the known HDAC inhihibitors, trichostatin-A, a fermentation product of Streptomyces, is effective in nanomolar concentrations and is the standard reference for this class of compounds. Other known HDAC inhibitors include short-chain fatty acids (such as valproic acid) and cyclic antibiotics such as apicidine. Currently, valproic acid is the only clinically applicable compound and is effective in the 0.5–1 mmol/L range, a concentration that is also considered therapeutic in the routine treatment of bipolar disorder. Although the potency of valproic acid on class I enzymes is 5 times higher,8,27 its comparatively low potency for HDAC inhibition may limit specificity in the clinical situation. Newer HDAC inhibitors with high specificity and low toxicity will be required to realize a therapeutic strategy from this approach to chromatin structure.
Alternatively, the targeted approach to chromatin structure allowing access to sequence-specific promoters, as mentioned earlier, entails the use of nuclear receptor ligands.32 LNRs function as epigenetic switches, and the last decade has seen the identification and characterization of approximately 40 such receptors in vertebrates.47 Many of these receptors have natural ligands that are small lipophilic molecules (e.g., steroids, vitamin D3, retinoic acid, products of lipid metabolism), some of which are generated in the cytosol. Several of these receptors have already been associated with known diseases, including diabetes, cancer and atherosclerosis, and retinoids have a wide range of clinical indications. Application of nuclear receptor pharmacology to psychiatric disorders will require development of ligands (both agonist and antagonist) with enhanced specificity and low behavioural toxicity. A hypothetical scenario is as follows. NURR1 is an “orphan” LNR (unknown ligand) that forms a heterodimer with retinoic X receptor (RXR), a well-characterized LNR activated by retinoids. Regulation of dopamine production in the midbrain depends on NURR1 function, and dopamine levels are depleted or reduced in NURR1 homozygous and heterozygous knockouts, respectively. The NURR1–RXR heterodimer could be activated by a retinoid ligand and might prove useful in the modulation of midbrain dopamine levels in patients with the deficit syndrome of schizophrenia.
Other approaches to altering chromatin structure include drugs that target and inactivate DNA methyltransferases (such as DNMT1, which is increased in the brains of patients with schizophrenia examined post mortem6). Compounds that either deplete DNMT1 levels or neutralize function are currently being examined for a potential role in cancer therapeutics. 48,49 Hydralazine and procainamide, used in the treatment of hypertension and cardiac arrythmias, respectively, have been shown to demethylate and reactivate gene expression in both in vitro and in vivo laboratory experiments, and hydralazine has been documented to demethylate DNA in malignant tissue from 2 human subjects.49 Clinical trials of these medications in psychiatric populations may prove their therapeutic efficacy.
Conclusions
An exciting development in this era of genomics is the excavation of a long-considered layer of gene regulation based on the sequence-independent modifications of the DNA strand and its surrounding proteins. The emerging paradigm is relatively free of the constraints and limitations inherent in attempting to change or manipulate the primary DNA code, and its cellular elements can be approached through conventional pharmacology. The recent discovery that valproic acid, used clinically for 3 decades in seizure disorder and bipolar disorder, has HDAC-inhibiting properties and can modify chromatin structure poses a whole new set of challenges and opportunities. Essentially, the relevant objective for the clinically oriented neuroscientist will be to characterize the behavioural implications of variant chromatin patterns in the neuron and to determine how to approach these structures using available or as-yet-undiscovered pharmacologic agents.
Acknowledgements
This work was supported in part by NIMH-069839 (R.P.S.), NIMH-62682 (D.R.G.), NIMH-062188 (A.G.) and NIMH-62090 (E.C.) from the National Institute of Mental Health.
Footnotes
Medical subject headings: neurons; chromatin; methylation; cognition; schizophrenia; bipolar disorder.
Competing interests: None declared by Drs. Grayson, Guidotti and Costa. Dr. Sharma has received speaker fees from Abbott Laboratories.
Contributors: All of the authors made substantial contributions to the conception and design of the study, the acquisition or analysis of the data, and the drafting or revision of the article. Each author gave final approval for the article to be published.
- Received July 29, 2004.
- Revision received February 16, 2005.
- Accepted February 22, 2005.
References

{kind=link}
Article tools



Related Articles
Cited By...
- No citing articles found.