

Abstract
Background: Alzheimer disease is a neurodegenerative disorder that progresses with marked interindividual clinical variability. We postulate the existence of endogenous molecules within the human brain exerting an antiaggregant activity that will prevent/slow Alzheimer disease progression.
Methods: We performed in silico studies to determine if the small endogenous molecules L-phosphoserine (L-PS) and 3-hydroxyanthranilic acid (3-HAA) could bind to the target region of β-amyloid responsible for protein misfolding. In vitro assays measured the antiaggregation effect of these molecules at varying concentrations.
Results: In silico studies demonstrated that L-PS and 3-HAA, both endogenous brain molecules, were capable of binding to the histidine13–histidine–glutamine–lysine16 (HHQK) region of β-amyloid involved in misfolding: these interactions were energetically favoured. The in vitro assays showed that both L-PS and 3-HAA were capable of inhibiting β-amyloid aggregation in a dose-dependent manner, with 3-HAA being more potent than L-PS.
Limitations: Studies were performed in silico and in vitro but not in vivo.
Conclusion: We successfully identified 2 endogenous brain molecules, L-PS and 3-HAA, that were capable of binding to the region of β-amyloid that leads to protein misfolding and neurotoxicity. Both L-PS and 3-HAA were able to inhibit β-amyloid aggregation in varying concentrations; levels of these compounds in the brain may impact their effectiveness in slowing/preventing β-amyloid aggregation.
Introduction
Alzheimer disease is a progressive, degenerative neurologic disorder for which there are no curative treatments. People with Alzheimer disease demonstrate characteristic pathological features, including β-amyloid plaques, tau neurofibrillary tangles and brain atrophy — collectively used to enable definitive postmortem diagnosis.1 The progression of Alzheimer disease varies tremendously among individuals from fast to extremely slow disease progression.2
The proposed causative agent in Alzheimer disease is the β-amyloid protein, ranging in length from 39 to 43 amino acids.3,4 A small, 4-residue segment within β-amyloid, histidine13–histidine–glutamine–lysine16 (HHQK; Appendix 1, Fig. S1, available at cma.ca/jpn), has been identified as a region that plays a mechanistic role in the conversion of β-amyloid from a nontoxic α-helical or random coil conformation to the β-sheet conformation that leads to aggregation and neurotoxicity.5,6 The aggregation of β-amyloid is a nucleation process whereby the misfolding of 1 protein initiates the misfolding of another, with the misfolded monomers forming into oligomeric species.7 The small, soluble oligomeric forms of β-amyloid are neurotoxic;8 thus, preventing their aggregation is therapeutically desirable. Once these oligomers have aggregated into fibrils and become sequestered in amyloid plaques, they are probably no longer toxic to the brain.4
Despite a common mechanistic pathway for the protein misfolding of Alzheimer disease, disease progression is highly variable among individuals. There are 2 main varieties of Alzheimer disease: sporadic cases with as-of-yet unknown triggers and familial Alzheimer disease, which is the early-onset, genetically triggered form.9 The incidence of sporadic Alzheimer disease increases as individuals age, with a 1 in 4 probability of Alzheimer disease developing after 85 years of age.9 The age of Alzheimer disease onset varies substantially, even among families with known genetic predisposition.10 There are specific genetic mutations that occur primarily with the presenilin 1 and 2 genes (PSEN1 and PSEN2) as well as the amyloid precursor protein (APP) gene, and it is these mutations that lead to alterations in β-amyloid protein load and generate β-amyloid length (which allows for the identification of β-amyloid as a pathogenic trigger).9 A variety of factors may play a role in why Alzheimer disease develops in certain individuals but not others (e.g., genetics, environmental factors, education level, socioeconomic status, life activities).10 Furthermore, the metabolic profile of the brain and immune factors may also correlate to the rate of cognitive decline among patients with Alzheimer disease, and there are 2 distinct streams of Alzheimer disease progression: rapid disease progression and slow disease progression.11
Along with variations in the rate of disease progression, there are also variations in how the disease manifests cognitively and behaviourally.12 These observed variations may be the result of differences in the accumulation of Alzheimer disease pathologies, or when these pathologies appear as the disease progresses.12 Despite all that is known about the heterogeneity of Alzheimer disease, there is still no clear understanding of why there are so many variations among patients. Why is it that some people may never get the disease, whereas others decline rapidly when Alzheimer disease develops?
To ascertain if individual neurochemical variability can account (at least in part) for the heterogeneity of the clinical course of Alzheimer disease, we pursued the notion of an endogenous anti-Alzheimer molecule (EAM). Accordingly, we sought to identify small molecules (molecular weight < 600 g/mol) in the brain that prevent or slow Alzheimer disease progression, specifically through inhibition of β-amyloid aggregation. Small molecules were selected because of their possible capacity to function as a drug-like platform and because of their potential to cross the blood–brain barrier if administered orally. To identify a putative EAM, we performed an initial in silico screening study of endogenous molecules interacting with β-amyloid followed by confirmatory in vitro assessments.
Methods
The HHQK region, which interacts with negatively charged glycosaminoglycans in the neuronal membrane, has been identified as a druggable domain within β-amyloid. If a compound can be identified that binds to this region, it could in principle prevent unwanted interactions that lead to protein misfolding.5,6 We devised an in silico screening strategy to identify compounds capable of binding to the HHQK domain. The HHQK receptor consists of 3 positively charged basic residues in a 1–2–4 arrangement within a tetrapeptide motif. Since binding to a single amino acid would not impart sufficient intermolecular binding selectivity, a molecule capable of binding to BBXB at either 2 or 3 of the “B” residues was required. This BBXB motif, where “B” is any of the amino acids with basic properties and X is any amino acid, has been identified as one that appears in many of the proteins affiliated with Alzheimer disease.13 Assuming an α-helical conformation and using a molecular mechanics energy minimized geometry of HHQK, the 1–2, 1–4 and 2–4 inter-residue side-chain charge separations are 7.1 Å, 8.5 Å and 13.2 Å, respectively. To establish energetically favourable intermolecular interactions with these cationic basic B-type residues, a preferred method is via an anionic group (forming a cationic-anionic interaction). Since we endeavoured to identify endogenous brain molecules that act as anti-Alzheimer therapeutics, we performed an in silico screen of 1100 endogenous small molecules of the human brain. We were interested in finding small molecules with negatively charged functional groups in a spatial orientation complementary to the cationic residues of the HHQK region. From this search we identified several “hits” — small molecules capable of fitting the appropriate dimensions and charge state for interacting with the HHQK region.
From this screen, 2 anionic metabolites formed within the human central nervous system were identified: L-phosphoserine (L-PS) and 3-hydroxyanthranilic acid (3-HAA). These 2 compounds were further submitted to more in-depth in silico studies and in vitro assays. For both L-PS and 3-HAA (Appendix 1, Fig. S2), we performed in silico optimizations with β-amyloid (in different conformations). Given the nature of these 2 molecules, we expanded the β-amyloid region of interest to residues Glu11-Lys16 (EVHHQK; Appendix 1, Fig. S3) as it provides additional opportunity for binding interactions. We also studied the inhibition of β-amyloid aggregation by both L-PS and 3-HAA in vitro using thioflavin T (ThT) and transmission electron microscopy (TEM) aggregation assays.
In silico simulations: L-phosphoserine
In silico geometry optimizations (via energy minimization calculations) of L-PS interacting with β-amyloid were computationally performed using the DREIDING2.21 force field, as implemented in the Cerius-2 operating suite.14,15 We used 6 different conformations of β-amyloid: 1AMB, 1AMC, 1AML, 1BA4, 1IYT and 2BP4 (as identified by their RCSB protein databank codes), which we then geometrically optimized at physiologic pH.16–21 A physiologically charged and geometrically optimized structure of L-PS was then constructed. Simulations were set up such that any 2 of the charged functional groups (carboxylate, phosphate and amino) were oriented 3.0 Å from any 2 of the charged amino acid side-chains in the EVHHQK region of β-amyloid for each of the 6 β-amyloid conformers. For each conformer, the 4 resulting systems with the lowest energies and multiple binding interactions were selected for detailed geometry optimization via energy minimization in an explicitly water solvated environment. Solution phase optimizations were performed with explicit solvation and periodic boundary conditions using the CHARM22 force field in the QUANTA program.22,23 We calculated the L-PS/β-amyloid binding energies for each system.
In silico simulations: 3-hydroxyanthranilic acid
We performed in silico simulations of 3-HAA interacting with β-amyloid using the CHARMM22 force field in the Molecular Operating Environment (MOE) software suite.24 The conformations of β-amyloid used for these calculations were 1AMB, 1AMC, 1AML, 1BA4, 1IYT and 1Z0Q.16–19,25 A geometry optimized structure of 3-HAA was oriented such that any 2 of the functional groups (hydroxyl, amino, carboxylate) and/or the aromatic ring were situated 3.0 Å from any 2 of the charged amino acids in the EVHHQK region of β-amyloid. A representative sample of systems from each conformer of β-amyloid examined was selected for optimization using explicit solvation. We performed solution phase optimizations with periodic boundary conditions using the CHARMM22 force field.
Confirmatory in vitro assays: thioflavin T
A ThT aggregation assay was used to quantify attenuation of β-amyloid aggregation by L-PS and 3-HAA. Thioflavin T, a benzothiazole dye, has a high affinity for protein with high β-sheet content and thus can be used to visualize β-amyloid aggregates. Unbound ThT has fluorescence excitation (λex) and emission (λem) wavelengths at 430nm and 342nm, respectively. Upon binding to aggregated β-amyloid, ThT undergoes a characteristic spectral shift (λex = 442 nm, λem = 482nm), which can be used to differentiate bound and unbound ThT.
We purchased β-amyloid 40 (> 95%) from AnaSpec and stored it at −80°C. Hexafluoroisopropanol (HFIP), 3-HAA, L-PS and materials for the tris(hydroxymethyl)aminomethane (Tris) buffers were obtained from Aldrich and were of the highest grade. All water used in the in vitro studies was micropore filtered and deionized. We pretreated β-amyloid 40 (1.0 mg) in a 1.5 mL microfuge tube with 1 mL of HFIP, and it was sonicated for 10 minutes to disassemble any preformed β-amyloid aggregates. We removed the HFIP with a stream of argon and dissolved the β-amyloid in Tris base (5.8 mL, 20 mM, pH ~10). The pH was adjusted to 7.4 with concentrated HCl (~10 μL), and we filtered the solution before usage with a syringe filter (0.2 μm). Pretreated β-amyloid 40 (40 μM in 20 mM Tris, pH 7.4) was diluted with an equal volume of 8 μM ThT in Tris (20 mM, pH 7.4, 300 mM NaCl). Aliquots of β-amyloid/ThT (200 μL) were added to wells of a black polystyrene 96-well plate, followed by 2 μL of L-PS or 3-HAA in dimethyl sulfoxide (DMSO). Incubations were performed in triplicate and contained 20 μM β-amyloid and various concentrations of L-PS or 3-HAA in 20 mM Tris, pH 7.4, 150 mM NaCl and 1% DMSO; DMSO alone served as a control, to ensure that any observed anti-aggregant effect was due solely to 3-HAA and L-PS. Plates were covered with clear polystyrene lids and incubated at 37°C in a Tecan Genios microplate reader. We obtained fluorescence readings (λex = 450 nm, λem = 480 nm) every 15 minutes after first shaking at high intensity for 15 seconds and then allowing to settle for 10 seconds before each reading. Active compounds attenuated the increase in fluorescence observed over time relative to the control samples.
Confirmatory in vitro assays: transmission electron microscopy
β-Amyloid 42 stock solution (40 μM in 20 mM Tris, pH 7.4) was incubated (37°C) in the absence and presence of 3-HAA or L-PS (100 μM). After 3 days, solutions were analyzed following the procedure of Cohen and colleagues26 for TEM analysis. A 10 μL sample was placed on a 400 mesh copper grid covered by carbon-stabilized Formvar film and allowed to stand for 1.5 minutes. Excess fluid was then removed, and the grids were negatively stained for 2 minutes with uranyl acetate (10 μL, 2% solution). Excess fluid was again removed, and the samples were viewed using an electron microscope operating at 80 kV.
Results
L-phosphoserine
Appendix 1, Table S1, summarizes the results of the in silico simulation of L-PS interacting with different conformations of β-amyloid; the final binding orientation of each system and the binding energies are presented. Only those systems that resulted in the formation of 2 or more energetically favourable binding interactions between L-PS and β-amyloid were included.
The results of the in silico calculations indicate that this small, endogenous molecule is capable of binding to the EVHHQK region of β-amyloid. The interactions between L-PS and the His13 and Lys16 residues are the most favoured for binding orientation, followed by those at His13 and His14, and those at Glu11 and His14. This indicates that L-PS can bind to β-amyloid at multiple sites within the target region. A successful binding interaction is depicted in Appendix 1, Figure S4.
A representative sample of interactions were selected for further optimization in a fully solvated environment. The results of the solution phase optimizations of L-PS with β-amyloid are summarized in Appendix 1, Table S2, with the initial and final binding orientations recorded. Any interactions occurring outside the EVHHQK region were listed under the “other” column, and the overall binding and electrostatic energies were measured for each system.
Of the 24 solution phase optimized systems, only 1 system did not result in binding interactions forming at 2 sites within EVHHQK. Analysis of the binding orientations suggested that interactions occurring at the His13 and Lys16 residues and the His13 and His14 residues were the most favoured, followed by those at Glu11 and Lys16. The measured binding energies of L-PS are more varied in the solvated environment, with no correlation between the number of binding interactions and the energetic favourability of the systems. The electrostatic energies of the observed systems suggest that all of these are favourable interactions.
The in vitro assays demonstrated that L-PS can inhibit β-amyloid aggregation. Figure 1 shows the results of the ThT assay of L-PS: as the concentration of L-PS increases, the observed fluorescence decreases. Figure 2 compares the amount of β-amyloid aggregation occurring after 24 hours in the presence or absence of L-PS. There is a significant difference in the amount of aggregation observed when L-PS is present.
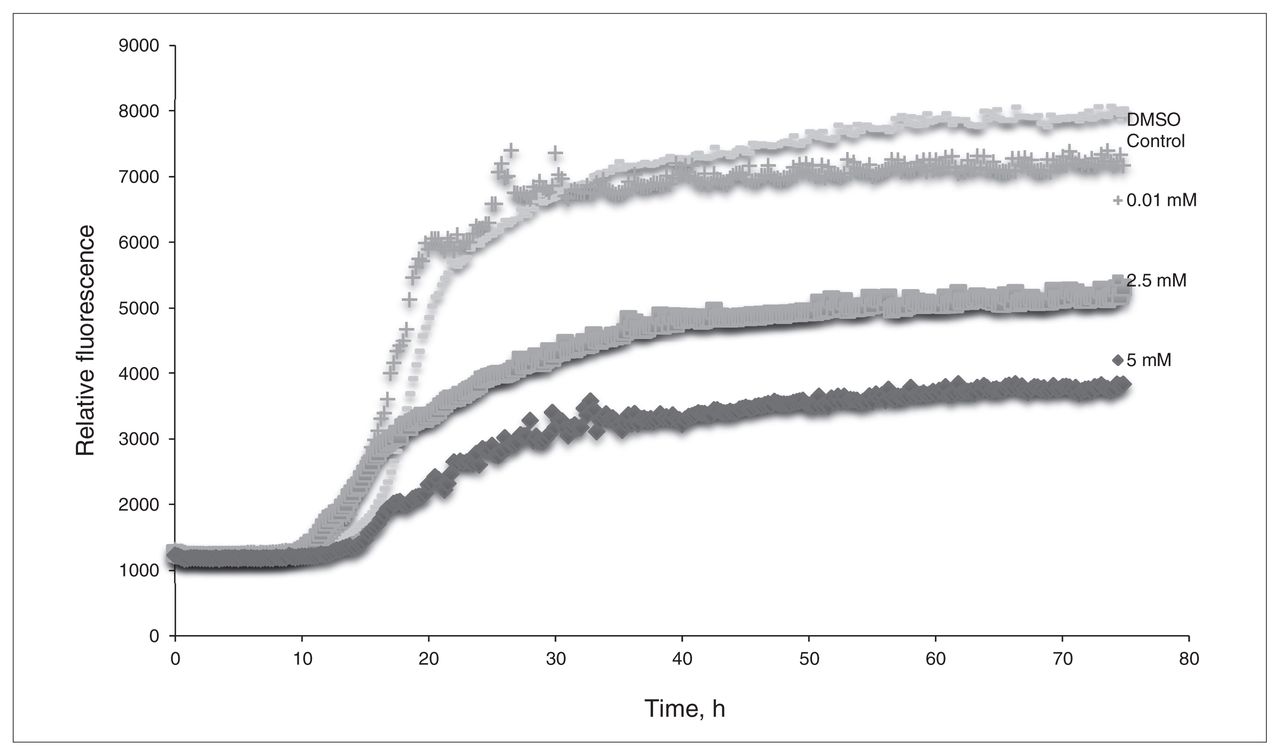
The thioflavin T assay of L-phosphoserine (L-PS). β-Amyloid was incubated with concentrations of L-PS of 0.01 mM, 2.5 mM and 5.0 mM. Dimethyl sulfoxide (DMSO) was used for a control sample.
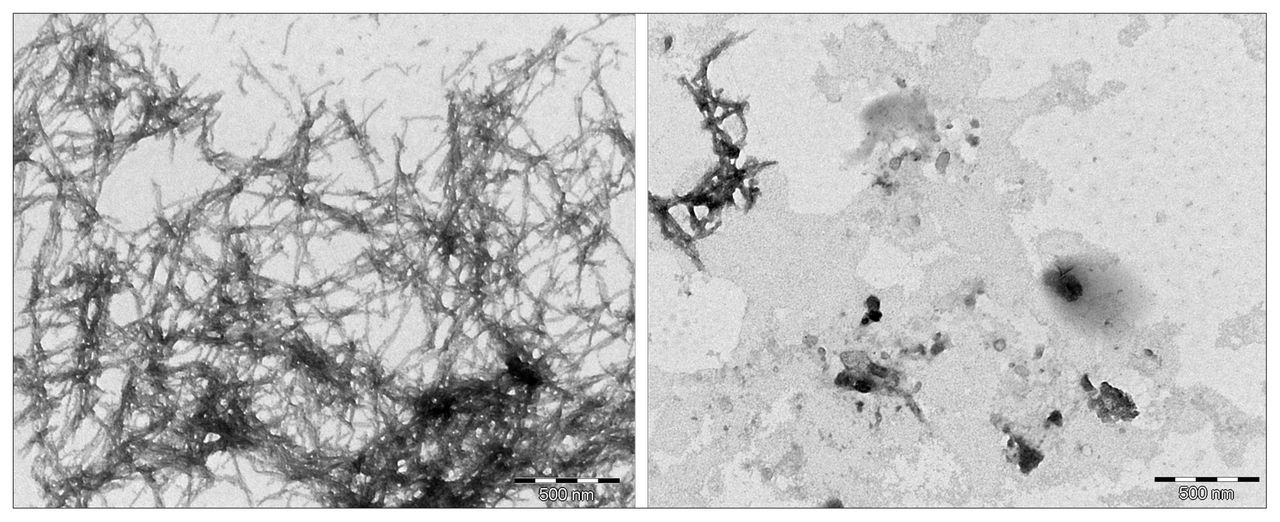
Transmission electron miscroscopy images of β-amyloid in dimethyl sulfoxide (left) versus β-amyloid with L-phosphoserine (right) after a 24-hour incubation period.
3-hydroxyanthranilic acid
The results of the in silico optimizations of 3-HAA interacting with different conformations of β-amyloid are summarized in Appendix 1, Table S3. The results are limited to those systems in which a minimum of 2 binding interactions have formed between 3-HAA and the protein. These results indicate that 3-HAA can interact with the EVHHQK region of β-amyloid, even when different conformational spaces are involved. There are 3 preferred binding orientations for 3-HAA to interact with 2 amino acid residues within EVH-HQK: His13–His14, Glu11–His14 and His13–Lys16. The measured binding energies of these systems were all favourable; an example of such an interaction is illustrated in Appendix 1, Figure S5.
From the preliminary optimizations, a representative sample of systems were selected from the interactions with 3-HAA and each of the different β-amyloid conformers to determine the effect that explicit water solvation would have on their binding. The results of the solution phase optimized systems are summarized in Appendix 1, Table S4, and show that even when explicit water molecules are present, 3-HAA is still capable of interacting with multiple amino acid residues within the EVHHQK region of β-amyloid. Of all the optimized systems, only 4 did not form more than 1 binding interaction with β-amyloid. Looking at binding interaction trends, systems where 3-HAA has bound to 2 amino acid residues within EVHHQK show preferred orientations of His13–His14 and Glu11–His14. The overall binding energies of these optimized systems are favourable. The ThT results in Figure 3 show a decrease in relative fluorescence as the concentration of 3-HAA increases. 3-hydroxyanthranilic acid clearly demonstrates a capacity to inhibit β-amyloid aggregation, and the concentration of 3-HAA required to inhibit β-amyloid aggregation is significantly less than that of L-PS. The TEM images in Figure 4 further demonstrate that 3-HAA significantly inhibits β-amyloid aggregation compared with a control sample. Only a few diffuse fibrils are present when incubation occurs with 3-HAA, relative to the control incubated with DMSO.
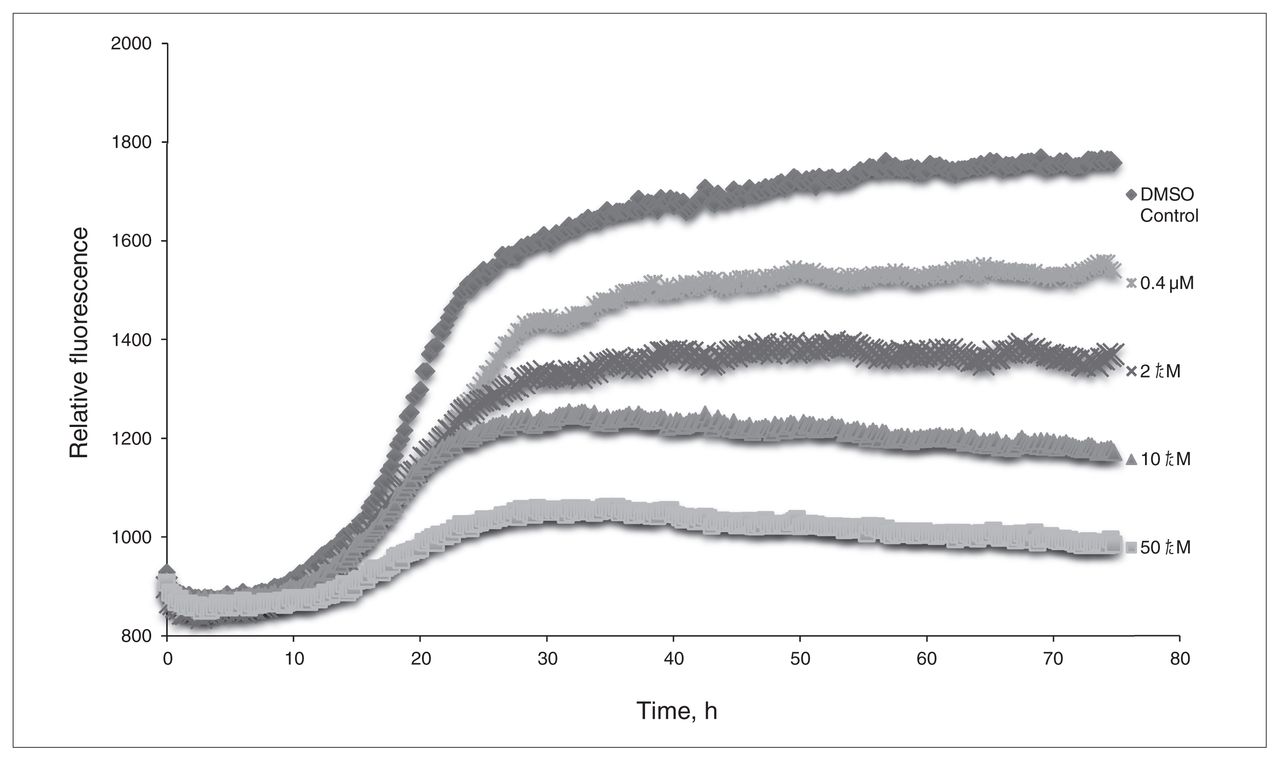
The thioflavin T assay of 3-hydroxyanthranilic acid (3-HAA). Relative fluorescence is measured versus time, with concentrations of 0.4 μM, 2.0 μM, 10.0 μM and 50.0 μM 3-HAA. Dimethyl sulfoxide (DMSO) was used as a control for β-amyloid aggregation.

Transmission electron miscroscopy images of β-amyloid aggregation after 24 hours in the absence (left) and presence (right) of 3-hydroxyanthranilic acid.
Discussion
In silico and in vitro methods have been successfully combined to suggest the existence of endogenous molecules within the human brain capable of inhibiting β-amyloid aggregation by binding to the EVHHQK region responsible for misfolding. Moreover, the 2 molecules identified, L-PS and 3-HAA, are small molecules and could conceivably be developed as drug discovery platforms.
L-phosphoserine
L-phosphoserine demonstrates a capacity to bind to β-amyloid in both in silico and in vitro studies. The in silico studies showed that L-PS was capable of binding to multiple amino acids within the EVHHQK region associated with the protein misfolding process. By binding to these specific amino acids, L-PS should block this highly charged region from interacting with negatively charged glycosaminoglycans. The favourable binding energies observed indicate that L-PS interacting with β-amyloid results in favourable, stable systems. Furthermore, the in vitro assays demonstrate that, although higher concentrations are needed to see a more significant inhibition of aggregation, the levels that normally exist within the human brain are enough to have a positive impact. Measurements of L-PS in the mammalian brain have proven to be challenging, and the levels of L-PS in the brain may also be impacted by interleukin-11, thus leading to conflicting results on the actual concentration.27,28 However, the only studies on the human brain have suggested that normal levels of L-PS in the brain are 0.3 mM, but an increase to levels of 1 mM can be seen in the brains of patients with Alzheimer disease.29 We were able to detect a decrease in β-amyloid aggregation at 0.01 mM; thus, the levels in the human brain should inhibit aggregation. The TEM images also clearly show that L-PS can prevent neurotoxic aggregation from occurring. These observations raise the possibility that the elevated level of L-PS occurring in the brains of people with Alzheimer disease may be a protective response (although others have suggested its role to be causative, rather than curative, because of a possible excitotoxic interaction of L-PS with glutamate receptors).29
3-hydroxyanthranilic acid
The in silico and in vitro assays revealed that 3-HAA has the capacity to bind to β-amyloid to prevent its aggregation. The ThT assay indicated that 3-HAA was capable of inhibiting β-amyloid aggregation at lower concentrations than those observed for L-PS and in a more significant manner. The TEM images have shown this β-amyloid antiaggregation effect to be significant as well, with only some diffuse aggregates forming in the same period during which significant aggregation occurs in a control sample. The results suggest that 3-HAA could be an even more effective antiaggregant than L-PS and that, as a metabolite of tryptophan, 3-HAA presents itself as a small, endogenous molecule target for further assessment. As with L-PS, elevated levels of 3-HAA and related metabolites have been found in postmortem brains with verified Alzheimer disease.30 These observations raise the possibility that the increased level of 3-HAA occurring in the brains of people with Alzheimer disease may in fact be a protective response (although others have suggested its role to be causative, rather than curative, because of a possible role of 3-HAA in mediating neurotoxic oxidative stress).30
Limitations
The main limitation of the present work is that these studies have only been performed in silico and in vitro. Although these methods are often representative of what would be seen in the human body, in vivo studies would allow us to better ascertain the success of these compounds if human trials were to be conducted.
Conclusion
The positive in silico results shown by L-PS and 3-HAA in binding to the EVHHQK region of β-amyloid in silico and inhibiting β-amyloid aggregation in vitro support the concept that there are endogenous molecules of the brain that can act as anti-Alzheimer therapeutics. These small molecules may also be used as platforms from which to develop even more potent amyloid antiaggregants in the search for a cure for Alzheimer disease. We have successfully identified small, endogenous molecules of the human brain that can inhibit β-amyloid aggregation by binding to the region involved in protein misfolding.
Acknowledgements
This research was funded by the Nova Scotia Health Research Foundation (A.R. Meek), the Gunn Family Studentship in Alzheimer’s Research (A.R. Meek), the Alzheimer’s Society of Canada (G.A. Simms) and Canadian Institutes for Health Research (operating grant to D.F. Weaver and studentship to G.A. Simms). The in vitro assays were performed by Todd Galloway and Rose Chen.
Footnotes
Competing interests: As above. D.F. Weaver also declares holding stock in Treventis Corporation.
Contributors: A. Meek and D.F. Weaver designed the study. A. Meek and G.A. Simms acquired the data, which all authors analyzed. All authors wrote and reviewed the article and approved the final version for publication.
- Received August 22, 2012.
- Revision received October 19, 2012.
- Revision received October 24, 2012.
- Accepted October 29, 2012.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.