

Abstract
A striking feature of psychosis is its heterogeneity. Presentations of psychosis vary from transient symptoms with no functional consequence in the general population to a tenacious illness at the other extreme, with a wide range of variable trajectories in between. Even among patients with schizophrenia, who are diagnosed on the basis of persistent deterioration, marked variation is seen in response to treatment, frequency of relapses and degree of eventual recovery. Existing theoretical accounts of psychosis focus almost exclusively on how symptoms are initially formed, with much less emphasis on explaining their variable course. In this review, I present an account that links several existing notions of the biology of psychosis with the variant clinical trajectories. My aim is to incorporate perspectives of systems neuroscience in a staging framework to explain the individual variations in illness course that follow the onset of psychosis.
Introduction
A striking feature of psychosis is the heterogeneity of its presentation. Psychosis-like experiences occur commonly in the general population, often without notable functional consequences. In general health care settings, psychotic symptoms that significantly affect one’s daily function occur in various disorders (e.g., delirium), often with full resolution. In psychiatric clinics, the course of psychosis varies from being a single, time-limited episode on one end of the spectrum to a tenacious illness at the other extreme, with a wide range of variable trajectories. Even among patients with schizophrenia, who are diagnosed on the basis of persistent deterioration, marked variation is present in response to treatment, frequency of relapses and degree of eventual recovery. Despite this, existing theoretical accounts of psychosis focus almost exclusively on how symptoms are initially formed, with much less emphasis on explaining their variable course. Thus, to date the focus of understanding the neurobiology of psychosis is largely at the symptom-formation level, rather than on the illness course in individuals. As a result, we continue to lack a physiologic framework to explain the wide range of variable outcomes that unravel to the point at which a patient experiences an initial episode of psychosis. In this article I attempt to construct an account that could link several existing notions of the biology of psychosis with the variant clinical trajectories. My aim is to put forward a thesis that could be invoked during a clinical dialogue with a concerned carer who is wondering why their loved one is presenting so differently from another patient attending the same treatment program.
First, I briefly review the concept of symptom resolution in psychosis and the evidence linking psychosis in general, and schizophrenia in particular, to cellular and systems-level brain connectivity. (The term “resolution” has been used to describe the reduction of symptoms to a level that is appreciable to the patient and care providers. The concept of durability of such resolution [i.e., remission] and its utility [i.e., recovery] are not the subjects of this review.) I will invoke existing ideas about the role of brain development, degeneration and plasticity to show how the concept of brain-network–level homeostasis can account for the varied course of psychosis. I also argue that the resolution of psychotic symptoms requires inherent homeostatic processes that, when aberrant, inhibit a fuller recovery. Finally, I highlight the aspects of psychotic illnesses that are not fully addressed by this framework and suggest future studies that are required to test the implications of the notions proposed here. I use the term “neural system stabilization” throughout this paper for the sake of simplicity, but this refers to the homeostatic process affecting all cellular constituents (i.e., glial cells and vasculature) that enable information transfer involving the entire brain.
Onset and resolution of psychosis
Psychotic disorders are clinically defined by and diagnosed based on the presence of delusions and hallucinations along-side deficits in processing speed, attention, verbal fluency, emotional expression, logicality and coherence of thought. These symptoms have a high probability of co-occurrence in some patients and demonstrate a variable degree of resolution after first presentation.
Several features of the natural course of psychosis call for explanation. 1,2 Isolated psychotic experiences such as voice-hearing and delusion-like ideas occur regularly among otherwise healthy individuals.3,4 Although these experiences are mostly transitory,5,6 the epidemiological risk factors for such experiences overlap substantially with those of psychotic disorders with conventionally poorer outcomes, such as schizophrenia. 3,4 People with psychosis-like experiences or the more tightly defined constructs of at-risk states or schizotypal disorder are at a higher risk of developing full-blown psychosis, but the majority of those who experience such transient psychotic states do not develop a psychotic episode;7,8 the risk of conversion peaks at 2 years and drops with longer follow-up periods.9
The onset of psychosis is often insidious or subacute in schizophrenia (53%10 to 70%11), but it can be florid and acute in many other psychotic disorders.12 The insidious prodromal stage in schizophrenia frequently presents with anxiety related to often numerous random coincidental associations and primitive perceptual aberrations,13–15 but at the peak of a fully evolved episode, psychosis is characterized by a limited number of stereotyped and fully formed delusions and hallucinations that tend to repeat.16 As psychosis evolves, patients often appear to add further elaborations to this limited set of delusions and hallucinations, rather than forming completely unrelated ideas (see Table 1 for first-person accounts17–19).
First-person accounts of delusional activity
A substantial number of patients with first-episode psychosis have only 1 episode.20–22 Globally, the incidence rates of such acute and transient psychotic disorders are consistently higher than the incidence of schizophrenia.23,24 Psychosis can resolve without treatment in some of these patients. 21,25 Longitudinal studies conducted before the advent of neuroleptics remind us that these numbers are large enough to not be dismissed.26 When appropriate treatment is started, a large number of first-episode patients show an early symptomatic response;27,28 such an early reduction in the severity of delusions and hallucinations predicts a favourable later outcome.29–31 Resolution of delusions does not usually involve “extinction” or “unlearning” the associations underlying psychotic beliefs; instead, it involves a gradual ability to detach from the pressure of the beliefs and perceptual abnormalities.15,32 While positive symptoms become less prominent with the course of illness, cognitive deficits and negative symptoms remain stable.33–35
Relapses can occur even in adequately medicated patients (primary relapse),36,37 but the rates of relapse are distinctly higher in those who discontinue treatment in the early stages of illness (interventional relapse).38,39 When relapses occur during the course of illness, irrespective of the duration of the intervening period of recovery, the same predictable set of symptoms tends to recur with each episode.40,41 Furthermore, unlike the first episode of psychosis, which is preceded by a long duration of prodromal symptoms and a gradual buildup of unusual experiences, relapses often occur without a similar insidious prodrome.36 With each relapse, treatment resistance becomes more likely, especially when the relapse occurs after discontinuation of antipsychotic therapy.36,39 Notwithstanding this phenomenon, a small proportion of patients achieve good function despite a high number of early relapses.42–44
Progressive structural changes of the brain in psychosis
One of the most consistent neuroimaging observations in patients with psychosis is a reduction in the amount of grey matter volume and thickness measured using MRI.45,46 These grey matter deficits are present even in the early stages of a patient’s life47,48 and are shared to some extent by their healthy siblings.49–51 Once the early psychotic symptoms come to the surface, these grey matter deficits appear to intensify,52,53 especially in the first few years,54 before slowing down.55 Some reports indicate a continuous but low level of ongoing grey matter reduction, even in much later stages.56,57 The extent of the grey matter deficits relates to both the severity of illness58– 60 and the degree of exposure to agents that are associated with relapses and functional disability.55,61,62 In addition, rather controversially, longitudinal grey matter reduction is more pronounced in those who have higher cumulative exposure to antipsychotic medications (with some differences between typical and atypical drugs).52,63,64 Nevertheless, at several brain regions, these changes appear to be reversible. Certain types of nondrug therapies appear to reverse or slow the structural changes in patients.65–68
Not every patient with psychosis shows progressive structural deficits, but in those who do show such grey matter changes, there seems to be a predilection for certain brain regions, including the superior temporal,53,60,69–75 lateral frontal, 71,72,75,76 insular52,60,70,73,77 and anterior cingulate70,71 cortex on a more consistent basis, followed by the thalamus,75 precuneus76 and inferior parietal75 and hippocampal74 regions somewhat less consistently. Interestingly, in the large-scale organization of correlated brain activity and structural connectivity, these regions constitute the so-called “hubs”78 or “rich clubs”79 in the human brain. Hubs and rich-club regions typically form the most connected nodes in the overall network architecture of the human brain and consequently show higher levels of overall activity.80,81 The connectional architecture and activity load experienced by these regions make them particularly vulnerable to structural damage in diffuse brain disorders such as schizophrenia.81,82
The widespread structural changes in psychotic disorders are often discussed in the context of either extended aberration in neurodevelopment or a limited form of neurodegeneration. While earlier theorists used the term degeneration to refer to the longitudinal changes in psychosis, it is now increasingly clear that neither a progressive neuronal loss nor a relentless clinical deterioration characteristic of true neurodegeneration occurs in psychotic disorders, leading to a preference for the term neuroprogression to denote post-onset brain changes.83 An overwhelming number of observations now exist that are interpreted as signs suggestive of aberrant neurodevelopment in schizophrenia.84–88 An equally strong line of argument exists for a limited form of neuroprogression. 89–92 Several attempts to bridge the 2 notions have been made in recent times.93–97 Most of the proposed compromises hinge on the notion that a healthy adult brain continues to develop and change in structure over time;98,99 a developmental aberration would continue to affect brain structure in adult life, thus explaining the neuroprogression in schizophrenia.100
Psychosis as a disorder of connectivity
The notion that psychosis is related to aberrant connectivity in the brain originated in the 19th century.101–104 The initial concept of disconnection was based on the manifest disconnection in thoughts, actions and behaviour seen in patients with psychosis. Various elegant theories later resurrected, refined and pinned this idea to the brain level, with the aid of postmortem and neuroimaging studies in the last 20 years.105–109 The 3 key pillars of the dysconnectivity hypothesis are (1) the reduction in neuropil, the tissue zone that normally houses a large number of neuronal synapses, observed in postmortem brains of patients with schizophrenia; 107,110,111 (2) abnormal increases and decreases in the correlation of activity among various brain regions (functional connectivity) measured using positron emission tomography, MRI, electroencephalography and magnetoencephalography studies;108,112 and (3) abnormal increases and decreases in the indices of white matter integrity (structural connectivity) measured using diffusion tensor imaging.113,114 These observations arise from different measurement tools used at different spatial and temporal scales and activity levels, but they are notably reconcilable at a whole brain (systems) level115,116 and speak to a reduction in the ability to transfer information within affected brains.
Although there are notable spatial variations in the patterns of resting-state functional connectivity in relation to psychosis, some patterns are now emerging consistently with improvised data-processing approaches.117,118 Functional hyperconnectivity, especially affecting the prefrontal cortex, is more pronounced during early stages of schizophrenia,119,120 relates to positive rather than the negative symptoms of the illness119,121,122 and normalizes to some extent with antipsychotic treatment.123,119 Such functional hyperconnectivity also results from external agents that typically induce psychotic symptoms. 124,125 Ketamine, an agent that produces nearly all of the core symptoms of schizophrenia in healthy humans, produces robust hyperconnectivity involving the prefrontal cortex.125–130 In particular, this resting-state hyperconnectivity involves a set of brain regions that constitute the default-mode network. 122,131,132 These regions characteristically show an elevated level of activity at rest and appear relatively deactivated when a person is engaged in task performance.133 In contrast to the prefrontal/default mode network hyperconnectivity in the early stages, a wider hypoconnected resting state is often noted in later stages of schizophrenia.112,120,126
Certain emerging observations provide clues as to the neural process that may underlie the hyperconnectivity between 2 brain regions seen in resting-state functional MRI. First, hyperconnectivity is often seen during the initial response to neuronal injury.134,135 Second, training and new learning in healthy brains results in an early increase in functional connectivity in relevant brain regions, along with hypoconnectivity.136,137 Even in the absence of a learning exercise, coordinated electrical/magnetic neural stimulation (plasticity-inducing paradigms) results in functional hyper-connectivity, indicating a Hebbian increase in neural communication at a synaptic level.138
The concept of neural system stabilization
The human brain can be regarded as a connected system that tolerates any faults by restoring itself. This concept has its roots in 3 broad theoretical notions: (1) Bernard and Cannon’s notion of “homeostasis” in biological systems;139,140 (2) the notion of self-organization in dynamic physical systems;141–143 and (3) the concept of fault tolerance employed in cybernetics, based on Dijkstra’s original network theory.144–146 In this section, I consider how these processes can scale up from synaptic to the macro-connectome level.
Homeostasis is a ubiquitous regulatory process that serves to maintain the function of a system at a set activity level, providing stability.147 The human brain is constantly bombarded by events and objects in the environment that evoke neuronal activity; in addition, constant spontaneous activity is also a feature of neuronal existence.148,149 Given the thousands of synapses that each neuron has with many other neurons, accidental coincidence of stochastic or stimulus-driven firing between 2 (or more) neurons is highly likely in this milieu.150 Hebbian rules of plasticity dictate that such coincidental spikes of activity will result in strengthening of the synaptic connectivity between the 2 neurons.151,152 But such an associative, input-specific learning process is often destabilizing to the neuronal ensemble, because it sets up the constituent neurons for either runaway hyperactivity or global silencing.147,153,154 If left unchecked, such a system can end in a hyperconnected or hypoconnected mode,155 neither of which is optimal for new learning or information transfer.156,157 The maintenance of both sparse functional connectivity and a steady baseline activity with low energy consumption are important for the status quo of the human brain.158
Several modes of homeostatic compensation operate to reduce the resulting instability;159,160 some of these involve functional rebalancing by stabilizing the firing rate of a neuron (intrinsic plasticity), tuning the inhibitory inputs (inhibitory plasticity) or down-weighting synapses (scaling). 161 In addition, structural alterations, such as changes in synaptic size, synaptic number and the dendritic spine structure (structural plasticity) either co-occur or serve as a second-level homeostatic mechanism.160,162,163 Reduced connectivity at the synaptic level is compensated for by an increase in the size164,165 and number of synapses, and reduced neuronal activity (e.g., via input deprivation) results in reduced synaptic elimination.166 These normal physiologic processes that regulate neuronal excitability or synaptic strength continuously degrade the absolute effect of synaptic coding that occurs with associative learning, but preserve the essential memory traces.153 Together, the homeostatic processes serve to maintain the overall excitatory and inhibitory balance in local neuronal ensembles that constitute the global brain connectome.167 This enables a connectome-wide system stabilization that facilitates optimal signal processing and learning.168 Such readiness is a prerequisite for continuous adaptation to one’s environment. 169 A similar self-restoring function has been described in various computational systems with distributed control, especially in the context of systemic fault tolerance that ensures the return of a perturbed system to its legitimate state in the service of global objectives.144,145
The notion of neural system stabilization in the global brain connectome refers to the scaled-up systemic effects of the homeostatic process operating at the synaptic level to maintain sparse connectivity and optimal activity, as summarized in Figure 1.
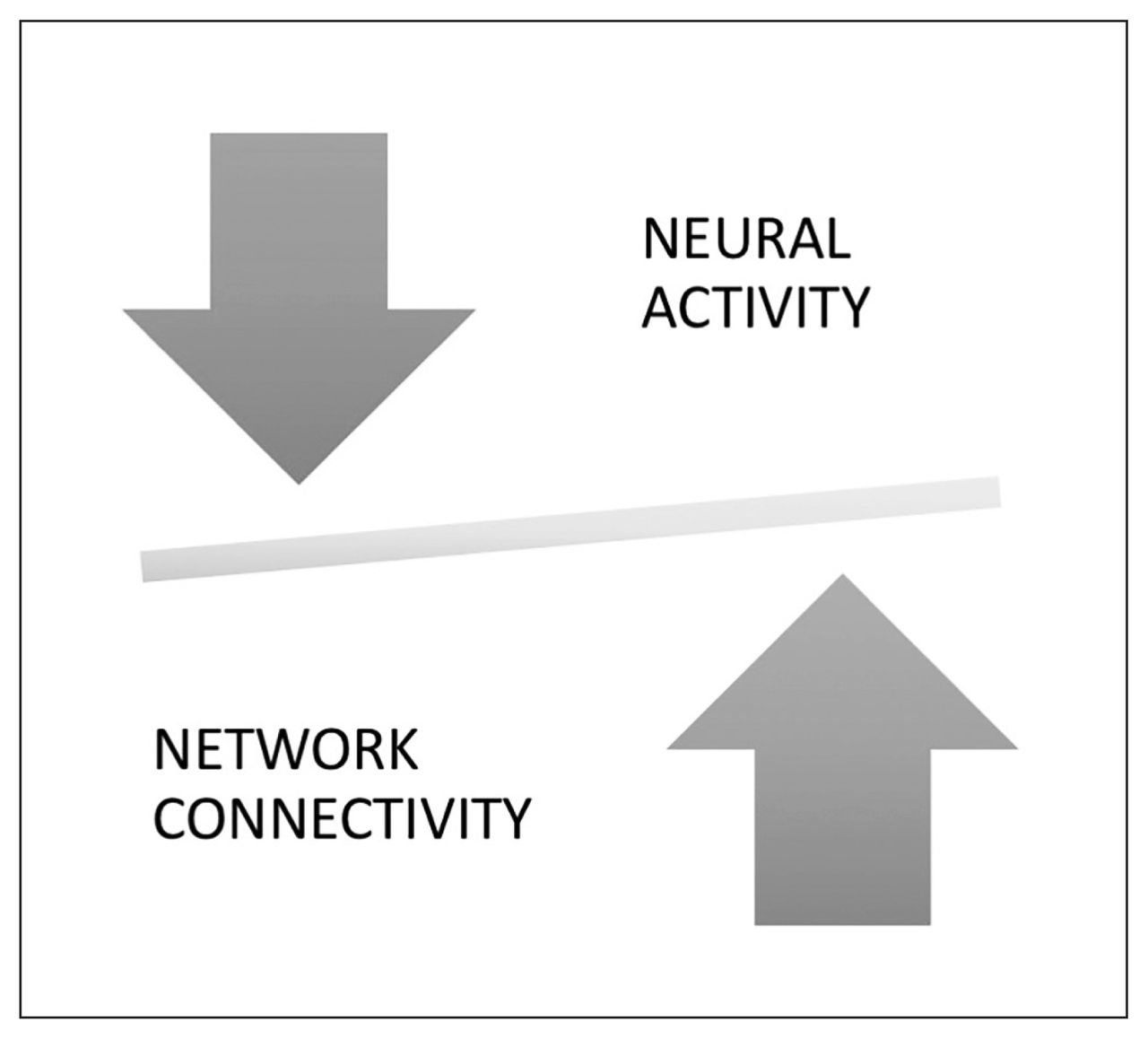
Illustration to reflect the balance between physiologic outputs of a neural unit (i.e., neuronal firing “activity”) and the social influence on the component unit (“connectivity”). If neuronal firing is excessive, the balance tilts, leading to engagement of homeostatic mechanisms that reduce the connectivity and restore the balance. Conversely, if synaptic strength/number (connectivity) is reduced, this triggers a compensatory increase in neuronal firing. This neural system stabilization helps to maintain topological homeostasis, likely characterized by a narrow range of “tuned states” of the brain connectome.
Psychosis and inefficient neural system stabilization
In the following section, I provide an account of how a disruption in neural system stabilization can result in the varied presentations of psychotic experiences. First, I propose that several factors can disrupt the stable pattern of timing-dependent coincidence detection at a neural level.
The first factor is intrinsic hyperactivity: a neural tissue with anomalously high frequency of activity can result in an increased probability of associative plasticity. Depending on the location of this activity, at an experiential level, we can speculate that people may experience brief sensory or cognitive disruptions of a fleeting nature. Several studies of people who were hallucinating have reported an elevated level of neural activity involving sensory cortices.170,171
The second factor is disturbances in the normal constraints on associative plasticity. A cardinal feature of Hebbian plasticity is dependence on the temporal order of coincidental neuronal activation;172 under certain circumstances, this temporal window can be prolonged, increasing the probability of formation of coincidences. For example, dopamine and substances that induce an excess release of dopamine could potentiate this mechanism.173
The third factor is failure of habituation: repeated presentation of the same stimulus elicits progressively smaller neuronal response.174 Disruptions in this habituation could prolong the state of evoked neuronal activity, increasing the probability of coincidences.175 A large body of electrophysiological studies points toward a habituation deficit in psychosis. 176–178 At an experiential level, we can speculate that people may report a lack of feeling of familiarity for events, again increasing ambiguity and uncertainty.175
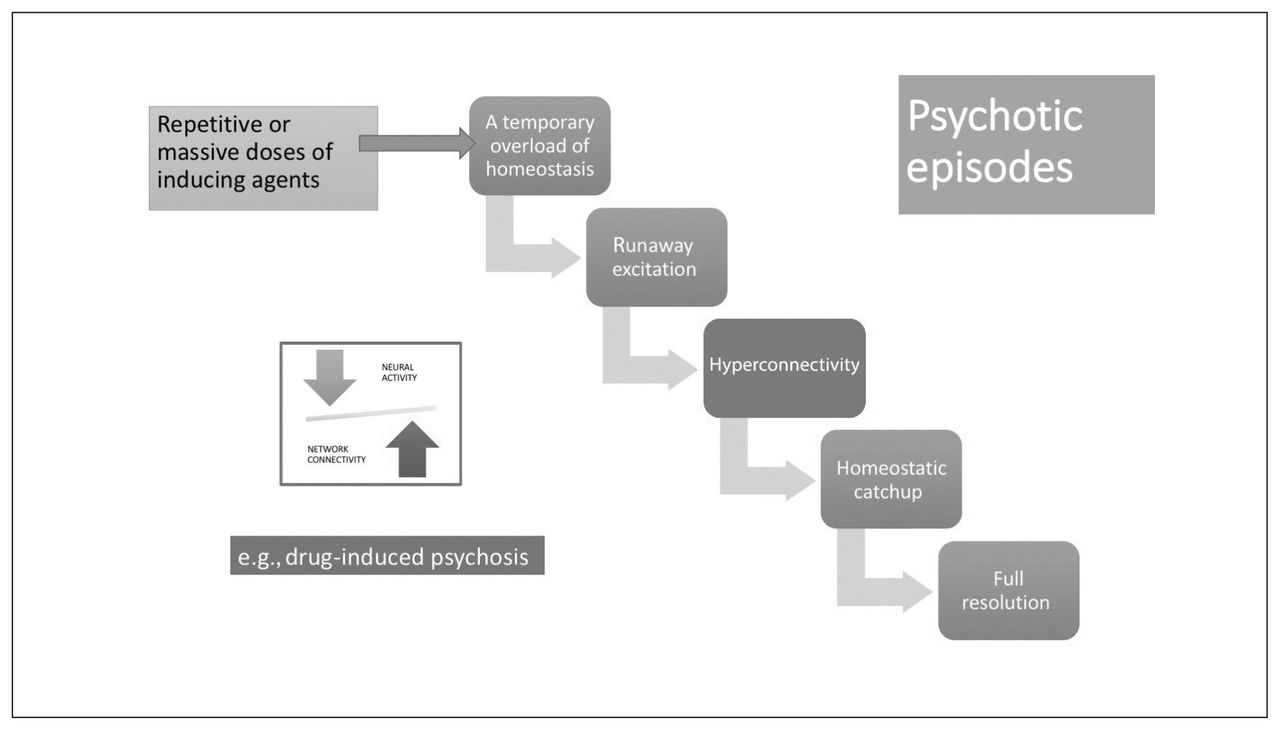
Although these aberrations clearly have the potential to generate psychotic experiences (Fig. 2), in each case, in the presence of an intact homeostatic plasticity, the synaptic coding will be weakened and eliminated. Thus, these disruptions can produce periodic psychosis-like experiences and prodromal features on their own but are insufficient to produce a psychotic episode. If agents that induce the above neural states are repeated frequently or occur in massive doses, then a temporary overload of homeostatic mechanisms can ensue, leading to a psychotic episode, although with a high probability of full resolution with or without treatment (Fig. 3). A likely example is the clinical presentation of drug-induced psychosis with full resolution. If this is the case, what is the necessary condition for the emergence of a schizophrenia-like illness?
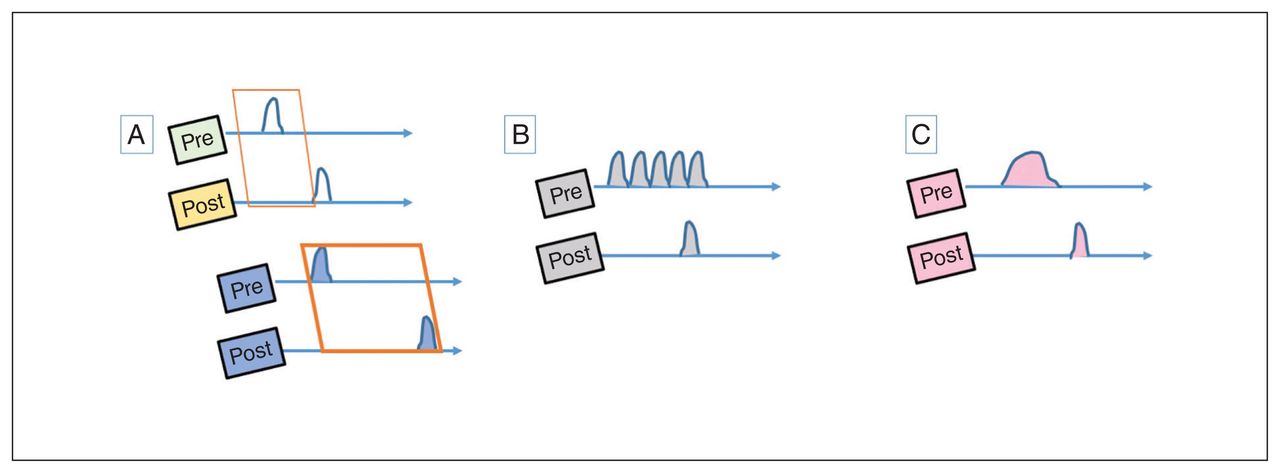
Anomalous associations and psychotic experiences. (A) Learning associations between 2 time-variable signals require tight temporal coordination (Hebbian window), shown as a narrow interval between the activation of pre- and postsynaptic neurons in the first illustration. This window can be prolonged in hyperdopaminergic states, as shown in the lower panel. (B) Anomalous bursts of presynaptic activity can lead to inadvertent Hebbian associations. (C) Failure of habituation may lead to prolonged states of evoked activity, increasing the probability of Hebbian associations.
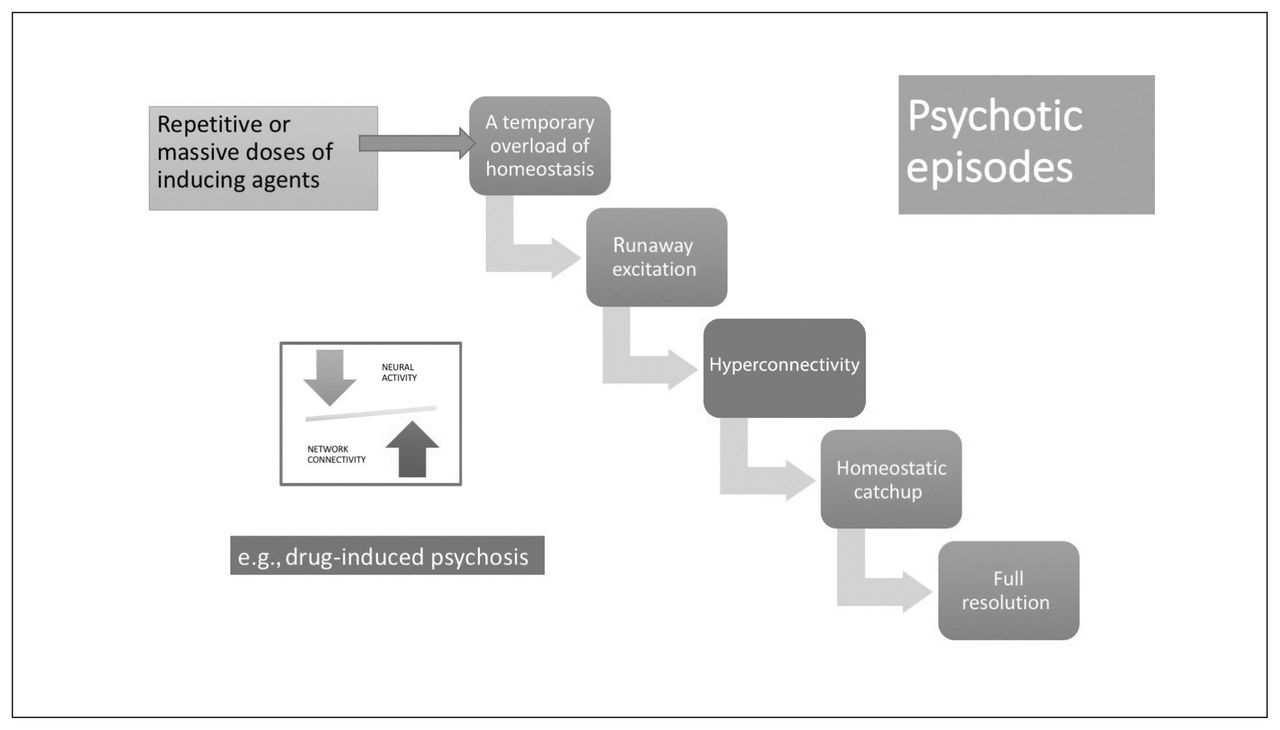
Stabilization lag in a psychotic episode. Psychotic episodes can occur after repetitive or massive doses of inducing agents through mechanisms shown in Figure 2, leading to a temporary overload of neural system stabilization. Provided that the cellular/topological system stabilization apparatus (homeostatic) is intact, these episodes can resolve fully.
I propose that in certain people, aberrant (nonstructural) homeostatic plasticity leads to a lack of resolution of coincidental associations. In such cases, synaptic strengthening accumulates (often insidiously) and the neuronal ensemble harbouring such coincidental associations eventually experiences a runaway excitation, forming a strong feed-forward loop that would quickly incorporate other correlations to the initial association (delusional elaboration characteristic of a psychotic episode), resulting in a state of indiscriminate hyper-connectivity in the proximate network space of the affected ensemble. In such circumstances, a recurrent, stereotyped temporal pattern is set in motion that can persist for an abnormally long period of time (“burnt into memory”155). This hyperconnectivity enables instantaneous recruitment of neuronal modules that are not normally involved in the processing of a given stimulus or task (“spreading activation”), producing the apparent hyperactivity in distant sites that is often reported in patients with psychosis. Topologically, the connectome now appears to be “subtly randomized” as sparsity in the connectivity is lost (as described by Rubinov and colleagues179). In the absence of homeostatic resetting, synaptic strengths of the affected neuronal core become fully saturated, precluding the formation of new associations that are required for extinction. Thus, once fully formed in an individual with inherent defects in homeostatic plasticity, the extinction of delusions becomes highly unlikely. Furthermore, the lack of neural system stabilization also reduces the connectome’s general readiness to process further inputs, thus reducing the speed of signal processing, expressed clinically as the negative symptom of psychomotor poverty. In addition, the suboptimal message-passing that occurs in an overloaded connectome (Fig. 4) can result in temporal delays in communication among modular brain units. This in turn can affect the temporal segregation required to separate self-generated from external mental content, 180–182 resulting in an aberrant sense of self. Thus, self-related disturbances and negative symptoms are more likely in those who have aberrations in neural system stabilization.

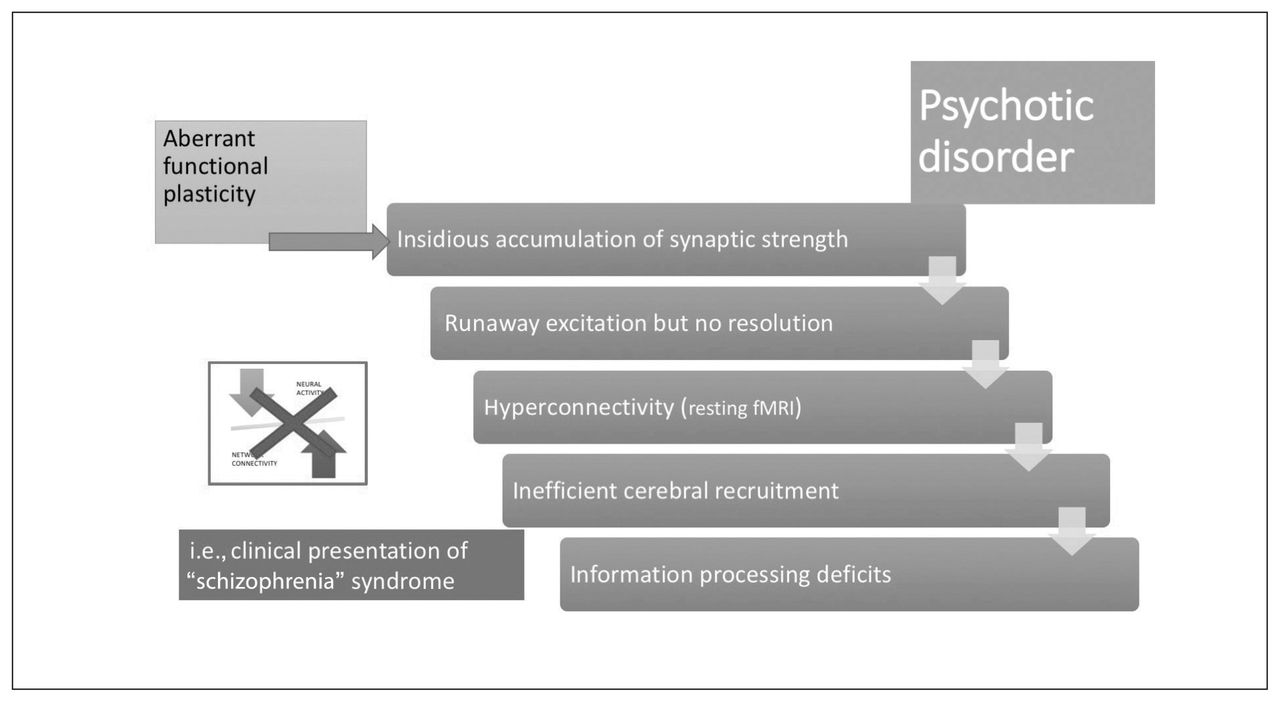
Stabilization shift in psychotic disorders. In those who have a predilection for aberrant functional plasticity, synaptic gains from associations accumulate over time, leading to runaway excitation in the neural network. The occurrence of this event may be brought forward by exposure to inducing agents, as indicated in Figure 3. In the absence of an intact neural system stabilization process, this results in a hyperconnected state for resting-state brain networks, with inefficient over-recruitment of task-processing regions. Subtle information-processing deficits that accompanied the predilection for aberrant functional plasticity now become more pronounced; the step change coincides with the first psychotic episode. The neural system stabilization mechanism now shifts from inefficient functional plasticity to a robust dependence on structural plasticity (i.e., spine reduction). fMRI = functional MRI.
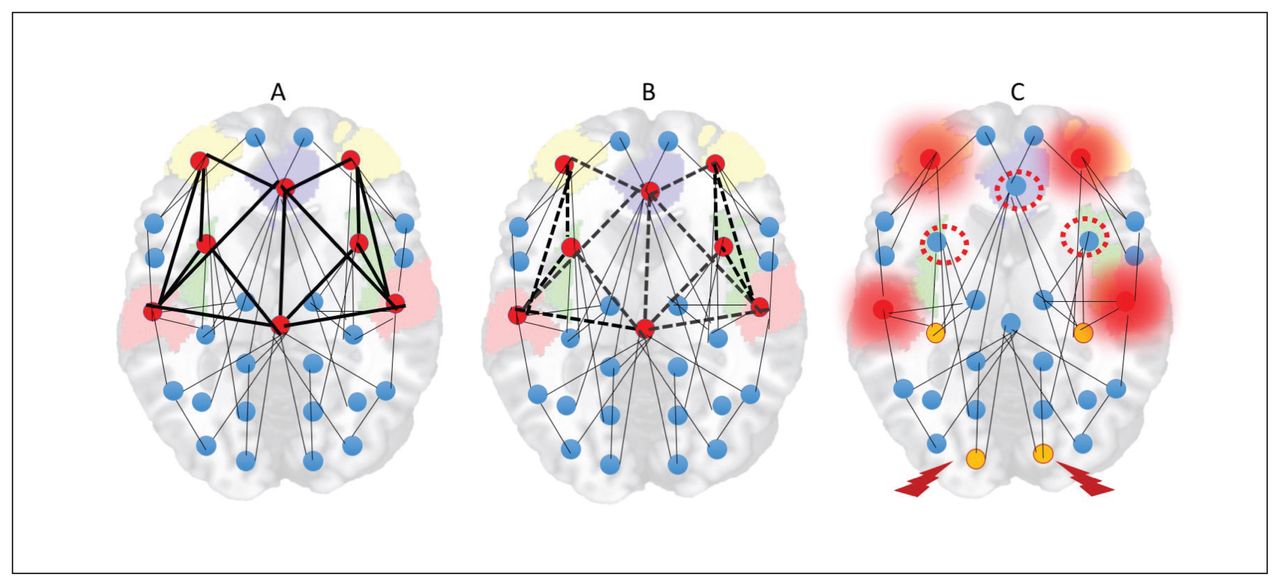
With the sustained and diffuse destabilization of the connectome that affects its sparse connectivity, the need for alternate homeostatic processes involving structural plasticity are triggered.162,183,184 Structural plasticity involves synaptic elimination and retraction of spines that aim to restore the sparse connectivity state that existed before the onset of psychosis. This process gradually eliminates excitatory dendritic synapses, with consequent neuropil reduction107,185 and progressive grey matter loss. Owing to the inherently higher activity levels and their propensity to be highly accessible to most of the aberrant neuronal ensembles, rich-club hubs of the human connectome (anterior cingulate cortex, insula, lateral prefrontal cortex, superior temporal gyrus and hippocampal regions) are most likely to be affected by this global retuning process.78,81,82,186 With time, this leads to “de-escalation” of hubs and restoration of sparse connectivity, albeit at the cost of increased segregation of functional modules and prolonged transit time in the network (Fig. 5).
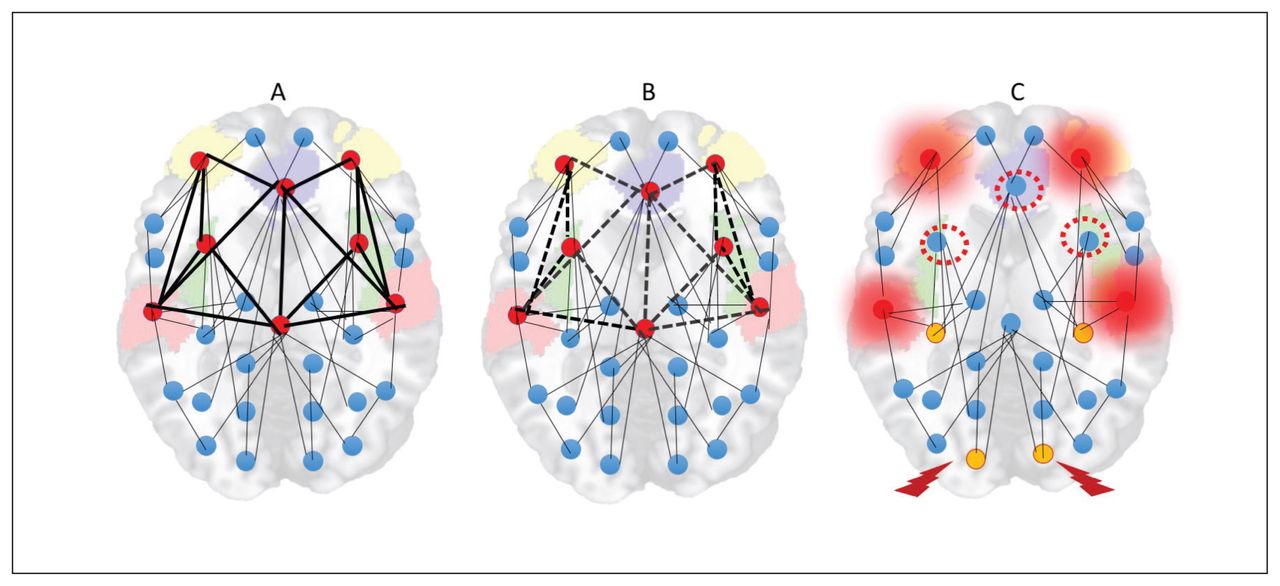
Hub de-escalation in psychotic disorders. (A) Rich-club hubs of the human connectome (anterior cingulate cortex, insula, lateral prefrontal cortex, superior temporal gyrus and hippocampal regions) have inherently high activity levels and higher topological proximity to any given brain region. (B) As a result, the pathways to and from these nodes are most likely to be the sites of dendritic spine reduction occurring in response to anomalous hyperconnectivity. (C) With time, this leads to “deescalation” of hubs (red-dotted circles), increased demands on remaining hubs (overloading effect, shown with a red halo) and emergence of peripheral hubs (yellow nodes). While this helps with restoration of sparse connectivity, it comes at the cost of increased segregation of functional modules (nodes with a thunderbolt sign less well connected to other hubs) and prolonged transit time in the network.
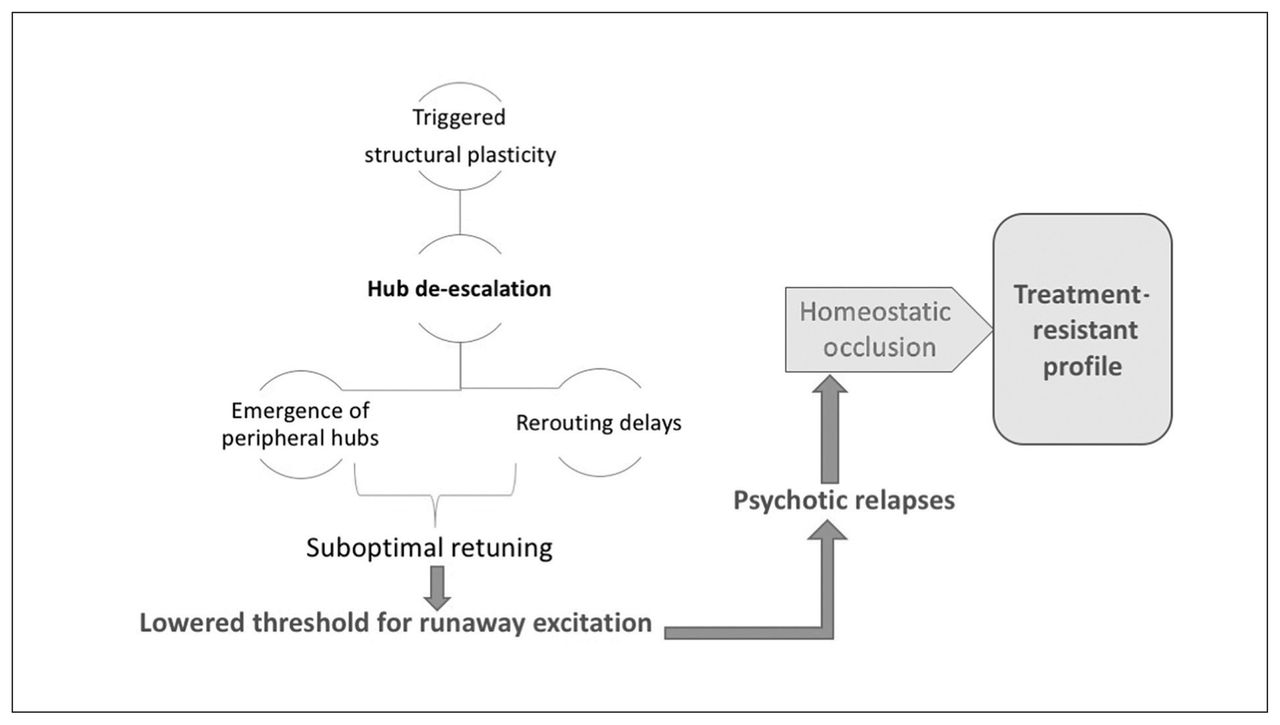
This structural homeostatic compensation eventually succeeds in abolishing the recurrent spreading of the avalanche of activity originating in anomalous neuronal ensembles. This comes at the cost of somewhat lower efficiency of information transfer, which becomes apparent whenever the demands on the system increase. Patients at this stage of illness exhibit a lower level of psychotic symptoms, but they continue to express negative symptoms (reduced speed of information processing, avoidance of social demands). One of the unintended consequences of hub/rich-club damage is the emergence of peripheral hubs, often located in unimodal cortices. These peripheral hubs, although they now have a higher degree of functional connectivity than other brain regions, lack the richness of hierarchical structure possessed by conventional core hubs. As a result, the flexibility of resource allocation is reduced at a global level, resulting in a reduction of system-wide plasticity and producing inefficient information transfer, especially when demands are placed on the networks. In addition, reduced overall connectivity increases the propensity of higher neural activity (Fig. 1), creating fertile soil for further relapses. Psychosis-inducing triggers continue to produce further episodes at this stage, although with reduced homeostatic reserve — both intrinsic and structural — even milder doses of inciting agents may now be sufficient to induce relapses.187,188 Furthermore, compensation through dendritic spine reduction reaches a critical point after repeated relapses with no further room for spine reduction, leading to a state of “homeostatic occlusion” (Fig. 6). In people who reach this stage, treating psychotic episodes becomes more difficult, taking longer and requiring higher doses, and some episodes will remain treatment-resistant.
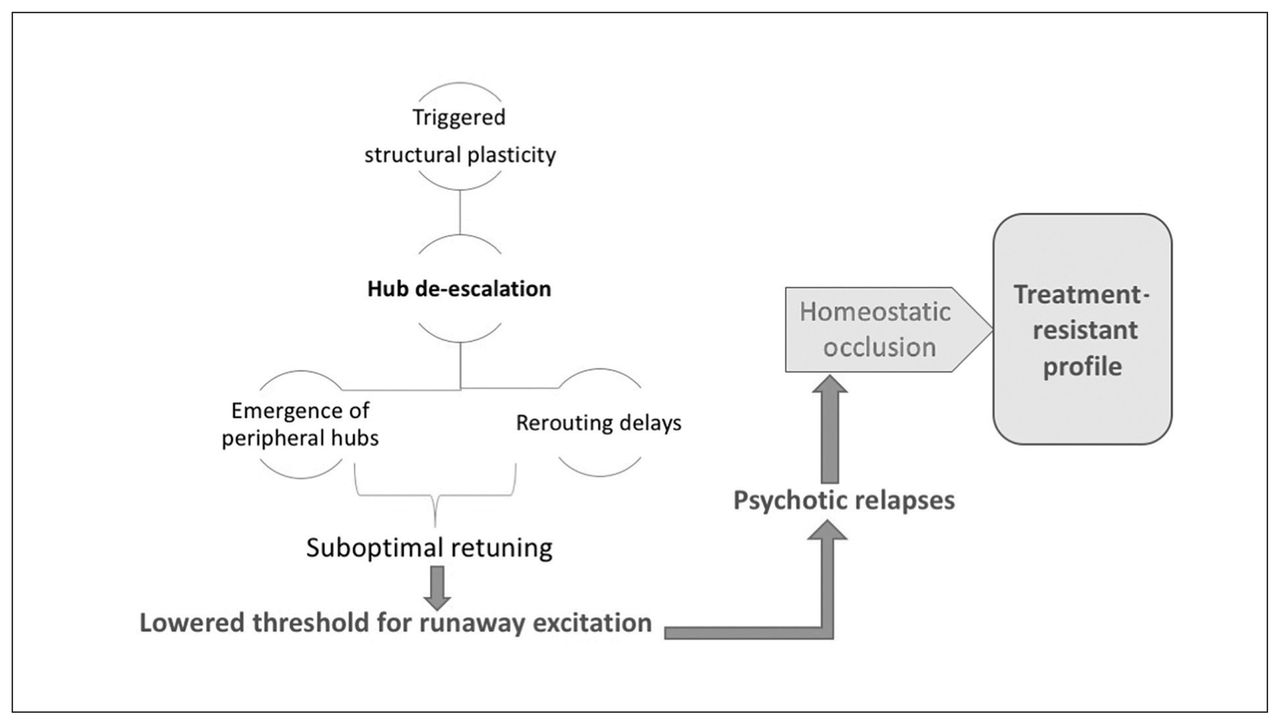
Saturation of system stabilization. Hub de-escalation results in a suboptimally retuned system that is prone to runaway excitation, even with milder doses of psychosis-inducing triggers. Recurrent relapses exhaust the structural plasticity through dendritic spine elimination, leading to a state of homeostatic occlusion. This is associated with a treatment-resistant state, in which interventions that act primarily by enhancing functional plasticity are no longer effective.
Many other agents that may worsen the outcome of established illness can directly induce dendritic spine elimination. 189 Drugs of abuse also alter the dynamics and microstructure of both dendrites and dendritic spines;190 chronic stress also promotes synaptic elimination.191 In addition, activity dependence of structural synaptic plasticity means that at least some of synaptic elimination could be secondary to lack of environmental stimulation.192 When the first psychotic episode occurs at a very early age, accompanying developmental processes synergistically hasten dendritic spine reduction, resulting in accelerated homeostatic occlusion and early emergence of treatment resistance.
Dopamine regulates the gain of N-methyl-d-aspartate (NMDA)–receptor-mediated Hebbian associative plasticity.193 Antipsychotics, by blocking D2 receptors, may act directly at the synaptic milieu to inhibit the associative learning process. They also facilitate the intrinsic functional homeostatic plasticity that counteracts the Hebbian potentiation, reducing the time taken for symptom resolution.194,195 In particular, typical neuroleptics result in the inhibition of long-term potentiation and spike-timing dependent plasticity.194 This effect rapidly reduces the runaway facilitation that occurs at the synaptic level during a psychotic episode, facilitating the resolution of the psychosis (early responders) and restoring the efficient sparse connectivity.196 Nevertheless, in patients with defective intrinsic homeostasis such rapid response may not occur; in addition, when response occurs eventually, discontinuation or dose reduction may carry a higher risk of relapse. Further, antipsychotics also increase oxidative stress, which may trigger or indirectly facilitate structural synaptic plasticity.197 With long-term use, certain antipsychotics (especially atypicals) may have a propensity to reduce synaptic elimination, halting or reducing the neuropil reduction that may occur in schizophrenia,198 and typical antipsychotics may alter synaptic proteins, facilitating dendritic spine regression and neuropil reduction.199 Critchlow and colleagues200 demonstrated a 59% increase in primary dendritic spine density in rat hippocampal neurons upon clozapine administration, while haloperidol had an opposing effect (see also Bringas and colleagues201). This suggests that clozapine may have specific effects on dendritic spines that may help restore structural plasticity, even in later stages of treatment resistance.
Excessive anomalous associations due to abnormalities in timing-dependent plasticity may result in psychotic experiences; if agents inducing such abnormalities are repeated frequently or in occur massive doses, then homeostatic mechanisms may experience temporary overload. This leads to a psychotic episode that can resolve with or without treatment, if the process of system stabilization is intact. In individuals with aberrant functional plasticity, inefficient cerebral recruitment and processing deficits occur, resulting in the clinical syndrome of psychotic disorders such as schizophrenia. In this state, hub de-escalation and suboptimal retuning of the global connectome provide fertile ground for relapses. With successive relapse, a state of homeostatic occlusion occurs, leading to a treatment-resistant profile.
Neural system stabilization, sex and developmental age
Brain development is characterized by critical time windows in which the system stability of the brain is notably affected. Such windows are characterized by an increase in structural homeostatic activity that helps to restore the stability. For example, adolescence is associated with crucial changes in the profile of NMDA receptors, which mediate functional synaptic plasticity (a subunit switch from NR2B to NR2A)202,203 and the dopamine receptor D2, which affects inhibitory plasticity.204 During this critical period, excitatory synapses are actively eliminated,204–206 increasing the inhibitory tone required for balanced brain activity. I posit that the emergence of psychosis is more common during late adolescence because excess demands are placed on structural plasticity at this time. Furthermore, homeostatic plasticity is the major mechanism of experience-dependent shaping of the developing brain. Any perturbation in this system—such as the one proposed here as a mechanism for psychosis—is bound to have a developmental effect that can present itself even before the onset of psychosis (e.g., disrupted asymmetry, 207 folding defects208). In particular, invocation of structural homeostatic plasticity could be related to an increased vulnerability of oxidative stress, which plays a crucial role in synaptic elimination.209,210 This may explain why obstetric complications such as neonatal hypoxia and consequent developmental defects are more likely to be seen in patients who later develop schizophrenia.211,212
Hebbian plasticity mechanisms appear to be modulated by sex-specific neurosteroids in animals.213,214 In particular, estradiol, allopregnanolone and related endogenous neuro-steroids serve to reduce intrinsic neuronal excitability through various mechanisms.215 Provided that such sex-specific mechanisms are preserved in people who later develop schizophrenia, the occurrence of the runaway excitation necessary for persistent psychosis and subsequent attempts contributing to grey matter loss must be less likely, taking longer to be established in women.216
Compatibility with emerging observations
Multiple lines of evidence support the various postulates that form the building blocks of inefficient neural system stabilization. A large body of evidence emerging from transcranial magnetic stimulation studies support the aberrations in plasticity mechanisms in schizophrenia.217–221 Graph-theory-based accounts using data from multiple neuroimaging modalities point to a subtle randomization of the topology of brain networks in schizophrenia despite the preservation of the sparse but efficient small-world structure of connectional architecture; 101,179,222,223 this effect is anticipated if the core hubs lose their prominence and peripheral hubs emerge in an attempt to restore the sparse connectivity.224
Further, excess microglial activity, which is suspected to contribute to grey matter reduction in schizophrenia,225 can also lead to homeostatic synaptic elimination.226
Genetic observations
Inefficient neural system stabilization, characterized by a tilt from functional homeostatic plasticity to structural plasticity, is compatible with many emerging observations that link inflammatory processes with psychosis. Genome-wide association studies now point to a dysregulation of synaptic glutamatergic function227 and inflammatory mediators in schizophrenia.228,229 In the major histocompatibility complex, the variations in the complement cascade appear to be the most prominent genetic abnormality associated with schizophrenia. 229 Complement cascade is dubbed the “masterful homeostatic regulator” of synaptic plasticity.230 Various products of this cascade are involved in tagging the appropriate synapses to be eliminated during normal development.231 Several other genetic loci implicated in psychotic disorders also point to mechanisms of homeostatic synaptic plasticity. 232 In addition to this polygenic risk, the less common but more penetrant copy number variations converge specifically “onto a coherent biological pathway at the synapse, with a specific role in plasticity.”233 In addition, genetic animal models that mimic various well-validated aspects of the schizophrenia phenotype converge on aberrant functional plasticity.234 Taken together, there is compelling evidence for the genetic basis of schizophrenia to converge on the regulation of glutamatergic synaptic plasticity.
γ-Aminobutyric acid and the glutamate system
A comprehensive review of postmortem studies suggested that the major contributor to grey matter volumetric loss in schizophrenia is synaptic regression involving glutamatergic excitatory synapses.110,235 On the other hand, functional deficits related to reduced GAD67 enzyme levels, rather than actual loss of parvalbumin-containing interneurons, are now well established in patients with schizophrenia.236 Deficiency in the GAD67 system does not appear to be a direct result of neurotransmitter dysfunction,237 but it may indicate the failure of a key functional homeostatic process that responds to excitatory synaptic activity;153,238–240 this weakening of inhibitory plasticity and subsequent connectome instability may serve to tilt the balance in favour of excitatory synaptic elimination in schizophrenia.241 This is consistent with our proposed model of inefficient neural system stabilization.
Glutamatergic concentration measured using magnetic resonance spectroscopy appears to be elevated during the early phase of illness but reduces during the course of illness, and this relates to grey matter loss at distal sites.75,242,243 The hallmarks of excitotoxic cell damage due to excess glutamate, if present, is yet to be demonstrated in schizophrenia, 244 but postmortem studies point to reduced glutamatergic dendritic spines and reduced biological coordination between glutamatergic signalling and synaptic structure. 245,246 The current model of inefficient system stabilization predicts that in early stages, higher levels of glutamate signal can be found in the loci of hyperconnectivity and anomalously increased neural activity, whereas lower levels of glutamate signal should accompany grey matter loss in schizophrenia, especially in later stages of illness, with excitatory synapses undergoing structural elimination. While several studies are in agreement,69,75,242,247,248 opposite results also exist.249 Cautious interpretation is required, because it is unclear how much of spectroscopic glutamate refers to synaptic activity.
The proposed notion of inefficient neural system stabilization in schizophrenia is in line with emerging literature on aberrant plasticity mechanisms, genetic observations about inflammation and aberrant glutamatergic signalling.
Compatibility with current theories
Dopamine occupies a central position in the current treatments of psychosis; midbrain dopaminergic pathways play a central role in network-level disruptions in schizophrenia.250 Dopaminergic receptors are concentrated on dendritic spines and play a key permissive role in modulating synaptic plasticity and associative learning.251 The hypothesis expounded in this review draws from existing accounts of associative learning, and so is broadly consistent with the models based on deficiencies in reinforcement learning and prediction error.252–258 Invoking disrupted plasticity as an explanation for psychotic disorders itself is not new. Stephan and colleagues posit that NMDA hypofunction is the key aspect of the plasticity defect seen in schizophrenia.259 One of the central tenets of this postulate is the ketamine model of schizophrenia. Ketamine mimics many acute symptoms of a psychotic episode, but it does not reproduce the features seen in established schizophrenia.260,261 Goto and colleagues262 highlighted the plasticity mechanisms of the prefrontal cortex and how they could be disrupted in schizophrenia. Keshavan and colleagues263 assembled a large body of evidence to argue that the positive symptoms of schizophrenia can be explained by hyperplasticity and negative symptoms by hypoplasticity, either being a compensatory response to the other. More recently, Forsyth and Lewis264 posited that the consequences of impaired synaptic plasticity during development explains the emergence of clinical symptoms of schizophrenia. System stabilization processes operating at the synaptic and macro-connectomic level are not invoked in the models proposed above.262–264
A recent proposition highlights the role of homeostatic compensations in the putative excitation–inhibition imbalance that starts with primary glutamatergic abnormality in schizophrenia.265 In contrast, the current model takes an agnostic view on the primacy of neurochemical abnormality. I propose that various disruptions in associative learning can invoke the earliest symptoms of psychosis, but that the core dysfunction in established schizophrenia lies in the homeostatic aspect of plasticity that is likely to involve primarily NMDA, but can also occur as a result of disruptions in other endogenous systems, such as the endocannabinoid pathways. By placing the emphasis on the homeostatic aspect of plasticity, the current hypothesis attempts to explain the continuum and course of schizophrenia rather than the acute phase of symptom appearance (Fig. 7). I also provide a framework that incorporates the emerging literature on connectomics, plasticity deficits, neuroprogression and inflammation and addresses distinct aspects of population-level variation in the course and outcome of primary psychotic disorders.
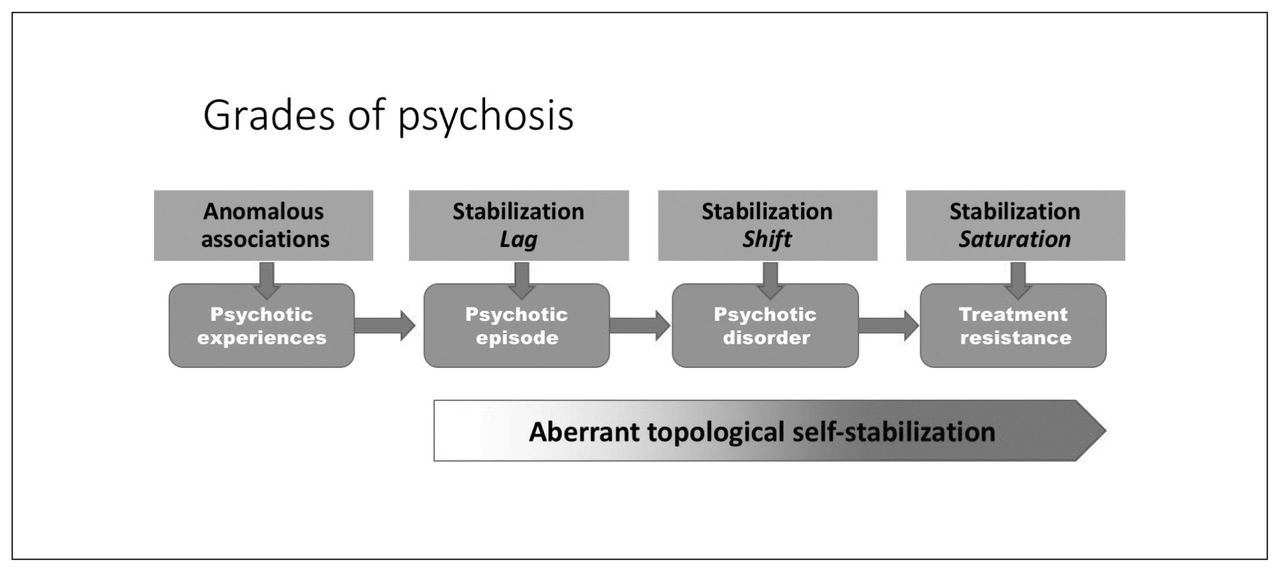
Neural system stabilization and grades of psychosis. The degree of impairment in the homeostatic process of topological neural system stabilization determines whether an episode of psychosis resolves fully, relapses repeatedly or fails to respond to currently prescribed interventions.
The role of the individual in neural system stabilization
Homeostatic processes operate at various levels to maintain the relative constancy of the regulated variables, and scale up from an intracellular to a societal milieu through hierarchical feedback processes.140 For example, intracellular sodium balance can be eventually traced up to the pre-dominance of human settlements close to water sources. Claude Bernard, one of the earliest proponents of the idea of homeostasis, maintains that the objective of this regulated constancy is to maintain the “harmonious whole.”266 This whole is often considered to be the “free and individual self”139 when studying biological systems, but it could refer to any of the higher levels of a self-organizing entity. Another important distinction between conventional homeostatic pathways and a self-organizing system is that an agent of control is not required to “sniff out” the perturbations in a self-organizing system.141
Does this mean an individual cannot have wilful control over the process of system stabilization? On the contrary: observations from clinical practice suggest that individuals have an intended control over the course of psychotic disorders. A strong sense of self,267 along with an internal locus of control of the illness,268 appear to be critical factors for recovery in psychotic disorders.
Damasio and Damasio argue that the feed-forward pathways of homeostasis give rise to conscious feeling states, which serve as “informative regulatory interfaces.”269 This mental aspect is an evolutionary advance that turns the individual organism into an agent of its own regulation. The “feeling state” generated by the connectome overload is likely to drive an individual to exert agentic guidance on system stabilization. Provided that the tools to correct (i.e., the physico-chemical apparatus for system stabilization) are accessible, individual efforts should hasten the restoration of the steady state. However, given the freedom of operation and societal influence, feeling-driven adjustments may not always be chosen to restore the perturbed system to its optimal state.269 This imperfection introduces another layer of heterogeneity in the prognostic trajectory of psychotic disorders.270
What is the nature of the feeling state that results from connectome overload? To answer the question, one must consider (1) the informative utility of the feeling state (i.e., the feeling must signal an adjustment that the organism is required to make in order to restore the system) and (2) the regulatory value (i.e., the feeling must serve to reduce further exposure to the triggers that perturb the system). It is tempting to speculate that secondary (or phasic271) negative symptoms (e.g., the feeling that nothing is pleasurable anymore; anhedonia)272 could signal the individual to reduce sensory, social and chemical stimuli that increase the likelihood of hyperconnectivity and further connectome overload.
The neural system stabilization theory proposed here allows for conscious agentic guidance of an individual in homeostatic processes, and raises interesting prospects regarding the wilful control of recovery in psychotic disorders (see Appendix 1 on the issue of insight and neural system stabilization, available at jpn.ca/180038-a1).
Predictions, limitations and further questions
Physiologic disruptions in neural processes related to learning based on temporal contingency are likely to be nonspecific to schizophrenia and will be shared not only by other psychotic disorders, but also by healthy people with psychosis-like experiences. But patients with schizophrenia are more likely to have specific disruptions in the system stabilization mechanisms that favour structural over functional plasticity. This vulnerability, if present from early life, could increase the risk of structural neurodevelopmental aberrations, various forms of intellectual and learning deficits even in the absence of psychosis (e.g., in siblings, or in premorbid stages273). But after the onset of symptoms, more extensive structural changes would be limited to those with inefficient system stabilization. As a result, morphometric changes may not be directly related to genetic liability for disease expression. But because tissue loss is related to neural plasticity, reversal of these structural deficits is possible.65 Identifying people at risk of more severe forms of psychosis and offering treatments that reduce or delay progressive synaptic changes at early stages may have a true disease-modifying effect. There are some promising venues in this respect.274–277 Established postonset grey matter loss is the sign of a “steady state,” albeit an imperfect one, that serves to induce further relapses. Reversal of deficits may require some disruption of this suboptimal steady state to enable the introduction of alternate means of neural system stability. To make therapeutic progress in established cases, it would be important to know how to safely carry out such topological restoration. Emerging neuromodulation approaches provide some promising leads.219
Given the heterogeneity of the currently accepted construct of schizophrenia, it is unlikely that any single theory could fit the multitude of observations pertaining to this disease. In particular, the notion of inefficient system stabilization does not offer causal explanations; it merely offers a mechanistic framework in which the operations of several causal agents could converge. In line with many other prevailing hypotheses, the current framework offers a better explanation for the course of positive symptoms; there is a greater degree of uncertainty surrounding the nature of relapse and the resolution of negative symptoms and thought disorder in schizophrenia. Further, the inefficient system stabilization model is inadequate for explaining the heterogeneity in symptoms of schizophrenia. Finally, the proposed theory does not provide a single index of a homeostatically regulated variable that can capture the inefficient system stabilization that can occur in all patients. Instead, it raises the suggestion that aberrations in various indicators representing the extant pathways of intrinsic plasticity and structural plasticity can be brought together to characterize a substantial number of patients with poor outcome.
Aberrations in homeostatic plasticity have been suspected in many neuropsychiatric disorders (see Appendix 1 on the issue of bipolar disorder and neural system stabilization). 155,232,262,278,279 Future studies aimed at discovering the aspects of this disruption that is specific to schizophrenia are required to fully understand the molecular pathways that can be targeted for treatment. If experimental models of disrupted homeostatic plasticity fail to mimic the true phenotype of schizophrenia despite the presence of conditions that disrupt associative plasticity, the theory proposed here could be refuted. To my knowledge, there is a striking lack of studies in patients who have recovered after a single psychotic episode, as well as longitudinal studies on connectome topology. Such studies are essential to test the premises of the neural system stabilization theory (Table 2; see Appendix 1 on the issue of insight and neural system stabilization).
Experimental approaches to test the neural system stabilization theory of schizophrenia
Conclusion
I speculate that psychosis, as seen in clinical practice, is not a disorder of any single neurotransmitter system or a particular brain network; instead, it is a disorder of cerebral acclimatization to new learning. An important aspect of this theory is that there is an intrinsic antipsychotic defence mechanism that promotes system stabilization in the human brain; this mechanism is built on the balancing act between neural activity and connectivity that is essential for learning statistical regularities in our immediate environment. Restoration of this intrinsic system stabilization can reverse many features of long-term psychotic illnesses. Critical timing of interventions, combined with approaches that provoke and redirect the pathways of neural network stability, may be required to realize this goal.
Acknowledgement
The author gratefully acknowledges Dr. Ross Norman for initial discussions; Prof. Tim Crow (Oxford) for reviewing this hypothesis and commenting on several aspects of this manuscript; and Dr. Ridha Joober and the attendees of CCNP Annual Meeting in Kingston, Ont., in June 2017, for stimulating questions that led to revisions of this work.
Footnotes
Funding: L. Palaniyappan’s work is supported by the Canadian Institute of Health Research (Foundation Grant 375104), the Bucke Family Fund and the Academic Medical Organization of South Western Ontario.
Competing interests: L. Palaniyappan reports personal fees from Janssen Canada, Otsuka Canada, SPMM Course Limited, UK, and the Canadian Psychiatric Association; book royalties from Oxford University Press; investigator-initiated educational grants from Sunovion, Janssen Canada and Otsuka Canada; and travel support from Boehringer Ingelheim and Magstim Limited, outside the submitted work. In the last 3 years, L. Palaniyappan and/or his spouse have held shares in Shire Pharmaceuticals and GlaxoSmithKline in their pension funds for values less than US$10 000. L. Palaniyappan joined the JPN editorial board in October 2019, after the current work was published online.
- Received March 13, 2018.
- Revision received February 7, 2019.
- Accepted March 5, 2019.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools


