

Abstract
Background: Phenotypic heterogeneity and complicated gene–environment interplay in etiology are among the primary factors that hinder the identification of genetic variants associated with cocaine use disorder.
Methods: To detect novel genetic variants associated with cocaine use disorder, we derived disease traits with reduced phenotypic heterogeneity using cluster analysis of a study sample (n = 9965). We then used these traits in genome-wide association tests, performed separately for 2070 African Americans and 1570 European Americans, using a new mixed model that accounted for the moderating effects of 5 childhood environmental factors. We used an independent sample (918 African Americans, 1382 European Americans) for replication.
Results: The cluster analysis yielded 5 cocaine use disorder subtypes, of which subtypes 4 (n = 3258) and 5 (n = 1916) comprised heavy cocaine users, had high heritability estimates (h2 = 0.66 and 0.64, respectively) and were used in association tests. Seven of the 13 identified genetic loci in the discovery phase were available in the replication sample. In African Americans, rs114492924 (discovery p = 1.23 × E−8), a single nucleotide polymorphism in LINC01411, was replicated in the replication sample (p = 3.63 × E−3). In a meta-analysis that combined the discovery and replication results, 3 loci in African Americans were significant genome-wide: rs10188036 in TRAK2 (p = 2.95 × E−8), del-1:15511771 in TMEM51 (p = 9.11 × E−10) and rs149843442 near LPHN2 (p = 3.50 × E−8).
Limitations: Lack of data prevented us from replicating 6 of the 13 identified loci.
Conclusion: Our results demonstrate the importance of considering phenotypic heterogeneity and gene–environment interplay in detecting genetic variations that contribute to cocaine use disorder, because new genetic loci have been identified using our novel analytic method.
Introduction
Cocaine is among the most widely abused illicit drugs in the United States.1 The National Survey on Drug Use and Health2 showed that in 2015, 0.7% of people aged 12 or older were cocaine users, an increase from 0.6% in 2014. Cocaine use is associated with serious health and social problems and is very costly to society, reflected in the fact that it is the illicit drug associated with the highest number of emergency department visits.3
Susceptibility to cocaine use disorder (CUD) includes a genetic component. Heritability of the 3 CUD-related traits — cocaine use, abuse and dependence — was estimated to be 0.39, 0.79 and 0.65, respectively, in female twins.4 Similar estimates in male twins were 0.61, 0.32 and 0.79, respectively.5 However, despite evidence of the heritability of CUD, there have been few efforts to identify specific genetic risk factors for the disorder.6 Several data sets with CUD traits have been used for genome-wide genotyping.7–9 To date, a single nucleotide polymorphism (SNP), rs2629540 mapping to FAM53B, has been associated genome-wide with CUD,9 an association for which consistent results in an animal model were later obtained.10
More than 10 biological processes, with more than 100 genes involved, may play roles in the etiopathology of substance use disorders.11 Variation in any of these genes — and indeed in other genes with an unrecognized relationship to these traits — could contribute to the development of a substance use disorder. Substance use disorders are heterogeneous and phenotypically and genetically complex, hindering the identification of specific genetic risk factors. In addition, multiple studies have shown that the genetic risk for developing a substance use disorder can be moderated by environmental factors such as stressful life events, neighbourhood stability, religiosity and peer drug use.12–18 Thus, the statistical power of genome-wide association studies (GWAS) to identify the genetic variation contributing to the risk of substance use disorders is limited by the extent to which environmental effects and phenotypic heterogeneity are unaccounted for.
In this study, we sought to identify genetic variants associated with CUD by conducting a GWAS using comparatively homogeneous subtypes and considering gene–environment interactions. We first performed multivariate cluster analysis using a discovery sample of 9965 participants for which we had a comprehensive clinical assessment. The analysis grouped cocaine users into homogeneous subgroups (i.e., subtypes) based on their clinical manifestations. We used the likelihood of membership in 2 highly heritable subtypes of CUD as traits in a subsequent GWAS and compared them with an ordinal trait derived by counting how many of the 11 diagnostic criteria for CUD in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) were present.19 We used a similar method to reduce phenotypic heterogeneity in a previous linkage study of cocaine dependence.20
Most of the participants were genotyped with genome-wide markers. We performed separate GWASs in 2070 African Americans (AAs) and 1570 European Americans (EAs) to identify SNPs associated with CUD. In the analyses, we also considered 5 childhood environmental factors, including nontraditional parental care, change in residence, traumatic experience, the presence of household drinking and illicit drug use, and the presence of household tobacco use. We used an independent sample of 918 AAs and 1382 EAs to replicate our findings, followed by a meta-analysis that combined the discovery and replication results.
All of our notable findings were observed in the AA population only. We identified more associations using the cluster-analysis-derived CUD traits than using the DSM-5 diagnostic criterion count. For all but 1 genetic locus, the genome-wide significant (GWS) findings were moderated by 1 of the 5 environmental factors and could not be detected with main effect association tests.
Methods
Participants and diagnostic procedures
A total of 11 000 participants were recruited for family-based (n = 2468 from 1047 small nuclear families) and case–control (n = 8532) genetic studies of opioid, cocaine or alcohol dependence. Participants were recruited at 5 sites in the eastern United States: Yale University School of Medicine (n = 5067), the University of Connecticut Health Center (n = 3765), the University of Pennsylvania School of Medicine (n = 1306), the Medical University of South Carolina (n = 607) and McLean Hospital (n = 255).
The institutional review board at each site approved all procedures; certificates of confidentiality were obtained from the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism, and all participants gave written informed consent to participate. Interviews were conducted using the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA),21,22 a computer-assisted interview that yields lifetime diagnoses for cocaine dependence and other major psychiatric traits using DSM-IV criteria.23 The reliability of the cocaine dependence diagnosis was excellent, with test–retest reliability of κ = 0.92 and inter-rater reliability of κ = 0.83.21
The reliability of the individual cocaine dependence criteria ranged from κ = 0.47 to 0.60.22 The SSADDA also covers criteria for cocaine abuse, 3 of which are included in the DSM-5 diagnosis of CUD. These 3 criteria, together with craving and the 7 DSM-IV criteria, were used to develop an ordinal DSM-5 CUD diagnostic trait.19 Moreover, a variety of other clinical features associated with cocaine use are queried in the SSADDA. The variables used in the subtyping (i.e., cluster analysis) procedure combined these clinical features. Finally, we included early childhood data in the analysis, obtained using the SSADDA environmental history section.
CUD subtypes
We used clinical data for 9965 participants (of the 11 000 total) — consisting of 2379 participants from 1099 small nuclear families and 7586 unrelated individuals — in the multivariate cluster analysis to develop CUD subtypes. We derived subtypes using 25 questions from the SSADDA cocaine section, which yielded 160 variables covering the following areas: (1) age of onset, frequency and intensity of cocaine use; (2) route of cocaine administration; (3) occurrence of psychosocial and medical consequences of cocaine use; (4) attempts to quit cocaine use; and (5) cocaine treatment history. We used 68 key variables from the 25 survey questions to generate clusters (see Bi and colleagues24 for a full description of the features used in the subtyping procedure). We used demographic and other substance use and psychiatric variables and disorders obtained from the SSADDA interview, together with heritability estimates, to characterize and evaluate the concurrent validity of the resultant clusters.
Differentiating the subtypes was a 2-phase process. Each phase comprised 3 consecutive steps: data reduction, cluster analysis and heritability estimation. We retained clusters from phase 1 with no cocaine-related features or a high heritability estimate (> 0.6) and merged the remaining clusters for use in the second phase. The second phase included only cocaine users as a means of refining the clusters. During each phase, we used multiple correspondence analysis (MCA)25,26 to reduce the large number of variables. We retained the top MCA dimensions that cumulatively explained 60% of the variance in each phase, leaving the top 25 MCA components in phase 1 and the top 41 in phase 2. We then used cluster analysis to group similar participants based on the retained dimensions. We first obtained 100 relatively small clusters using the K-medoids clustering method.27,28 These acted as intermediate clusters, to which we applied the agglomerative hierarchical clustering method,29,30 merging them into 5 clusters using Wald’s aggregation criterion and the Euclidean distance to compute the similarities between each pairs of intermediate clusters for merging. We determined the final number of clusters by manually inspecting the clinical characteristics of the resultant clusters at the different levels (from the 6-cluster level to 3-cluster level) of the clustering dendrogram. We constructed a probabilistic classifier using logistic regression and the 68 variables from the cluster analysis to separate participants in each cluster from those in other clusters. The classification probability provided an estimated likelihood of subtype membership for each participant, a continuous outcome variable of 0 to 1 that reflected the membership likelihood in each subtype. This likelihood measure reflects the phenotypic heterogeneity among the individuals in a cluster and those outside the cluster, providing a quantitative trait related to CUD rather than a qualitative trait. We estimated narrow-sense heritability for this quantitative trait for each subtype (cluster) using the “polygenic” function in the software package Sequential Oligogenic Linkage Analysis Routine (SOLAR)30 and the pedigrees of the sample participants.
We performed GWAS using the 2 most severe and highly heritable subtypes to maximize the likelihood of finding genetic associations. For participants in the GWAS or the replication analysis who were not included in the cluster analysis, we used the constructed classifier to calculate the trait value (i.e., the likelihood of the participants’ membership in a subtype based on the clinical variables).
Environmental measures
There are 33 questions in the SSADDA environment section that cover information on 11 major childhood environmental factors (e.g., change in residence, experience of violence, experience of sexual abuse, household drinking and illicit drug use). For the majority of the environmental factors, less than 23% of individuals in both the GWAS and replication samples were exposed (i.e., had a positive response). Because the small sample size yielded limited power to detect an effect, we limited the analysis to the 4 environmental factors for which the number of exposed participants exceeded 55% in both samples. There were also 3 important SSADDA environmental variables. Although they were endorsed by a very limited number of genotyped participants, we created a composite factor consisting of these 3 variables to increase the power to detect genetic association.
We derived a binary variable for each environmental factor. We evaluated “nontraditional parental care” based on the question, “Who was the main person taking care of you when you were growing up (before age 18)?” and considered the variable positive when the answer was anything other than “both mother and father.” These other responses included “mother or father plus step-parent,” “mother,” “father,” “grandmother,” “older brother or sister,” “other relative,” “foster parent” and “adoptive parent.” Nontraditional parental care defined here is closely related to parental separation, which has been linked to an increased likelihood of substance abuse or dependence, including illicit drug abuse.31,32
We evaluated “Change in residence” based on the question, “How many times did you move by age 13?” and considered the variable negative when the answer was “none.” Frequent residence change increases the chance of social disruption and exposure to diverse social norms and neighbourhoods. Social norms have been shown to have a predictive association with substance use.33,34 Neighbourhood instability has long been linked to drug use and dealing, as well as individual delinquency.35–37
We created a composite factor — “traumatic experience” — based on responses to 3 SSADDA questions: “Did you ever witness or experience a violent crime, like a shooting or a rape, by age 13?,” “By the time you were age 13, were you ever sexually abused?,” and “By the time you were age 13, were you ever beaten by an adult so badly that you needed medical care or had marks on your body that lasted for more than 30 days?” We considered this composite factor to be positive if the response to any of the 3 questions was yes. Childhood experiences of violent crime and sexual and physical abuse have been linked to an increased risk in adults of using substances, including cocaine.38,39
We evaluated “household drinking and illicit drug use” based on the question, “Were you ever aware of adults in your household drinking enough to get drunk, or using drugs or alcohol, by the time you were 13?” and considered the variable positive when the answer was yes. There is substantial evidence showing that a family history of drinking or illicit drug use predicts similar behaviours in offspring or siblings.40,41
Finally, we evaluated “household tobacco use” based on the question, “Were any members of your household regular cigarette smokers by the time you were 13?” and considered the variable positive when the answer was yes. Although a family history of smoking has not been directly linked to the use of cocaine in offspring, it does have a predictive effect on cannabis use in offspring.35,42
Genotyping and quality control
The sample used in the GWAS discovery phase was selected from among 5540 participants following quality control,9,43,44 and included 3640 individuals (2070 AAs; 1570 EAs) who had been exposed to cocaine and for whom we had data on the environmental variables. The replication sample included 2300 individuals with cocaine exposure (918 AAs, 1382 EAs) who were a subset of a larger sample that was genotyped using an exome microarray (n = 3675, following quality control).
The GWAS data were obtained using the Illumina HumanOmni1-Quad v1.0 microarray, which contains 988 306 autosomal SNPs, at the Center for Inherited Disease Research and the Yale Center for Genome Analysis. Genotypes were called using GenomeStudio software v2011.1 and genotyping module v1.8.4 (Illumina). Individuals in the exome microarray were genotyped with the Infinium CoreExome-24 Kit (Illumina), and genotypes were called using GenCall software ( Illumina). After a series of quality-control steps, the data set included 5540 individuals and 889 659 SNPs with GWAS data, and 3625 individuals and 261 746 SNPs with exome microarray data for imputation. Imputation was performed with IMPUTE245 using the 1000 Genomes reference panel (www.1000genomes.org/; released March 2012).46 For both discovery and replication samples, a total of 47 104 916 variants were imputed. We limited the association analysis to imputed variants with an imputation quality (INFO) score of r2 > 0.8.
To verify and correct any misclassification of self-reported race, we compared the GWAS (and exome microarray) data from all participants with genotypes from the HapMap 3 reference populations CEU, YRI and CHB. We conducted principal components analysis in the discovery and replication samples separately. To choose variants for the principal components analysis for each sample set, we first filtered out variants with a minor allele frequency less than 3% and INFO score r2 < 0.99. Then we identified SNPs that were common in our data sets and in the HapMap panel. Finally, we pruned SNPs in close linkage disequilibrium (i.e., r2 < 0.80%). This left 265 043 SNPs in the GWAS data set and 53 450 SNPs in the exome microarray data set for the principal components analysis. In both data sets, the first principal component distinguished AAs and EAs, aligning well with self-reported race, with few mismatching cases (Appendix 1, Figures S1 and S2, available at jpn.ca/180098-a1). We used the K-means clustering method in the first principal component dimension to partition the samples in both data sets into AAs and EAs (Appendix 1, Table S1). All subsequent association analyses were conducted separately by population group, with the first 3 principal components used to correct for residual population stratification.
We estimated the genetic relationship among participants separately for the discovery and replication samples using the linkage-disequilibrium adjusted kinships software,47 which takes into account linkage disequilibrium among the genetic variants. For both sample sets, only variants with a minor allele frequency of 3% or greater and INFO score r2 ≥ 0.99 were used in the genetic relationship estimation. There were 3 104 531 and 604 884 such variants in the GWAS and exome microarray data sets, respectively.
Statistical analysis
Traditionally, to identify gene–environment interaction, the following multiple regression model is used:
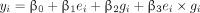
where i indicates the ith participant in the data; ei and gi represent the environmental factor value and genotype of interest of the ith participant, respectively; ei × gi is the interaction of the 2; and the βs are the model parameters.48 This model can also consider covariate effects and model residual, which we omitted to simplify the subsequent notations. An estimated value of β3 that differs significantly from zero indicates that e and g have an interactive effect on y. Our goal was to test whether g had an influence on y when a moderating effect of e was taken into account. Therefore, we excluded the g term and adopted the following model:
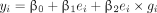 (1)
(1)In our data, all environmental factors were binary (i.e., ei took a value of 1 or 0). Plugging 0 and 1 into model 1 to replace ei, we had:
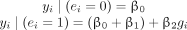
So, a significant nonzero β2 would indicate that g had an effect on y in the presence of e.
In our analyses, we accounted for both fixed effects from several covariates (e.g., age and sex) and a random effect from genetic relationship among individuals using a mixed model adapted from model 1 as follows:
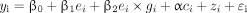 (2)
(2)where ci was the vector of values that the ith participant had for the covariates of interest; α was the vector that contained model coefficients of these covariates; zi represented the random genetic effect; and ɛi was the model residual. To answer the question of whether the genetic association identified with model 2 was due only to the effect of the variant itself (i.e., with no effect of the environmental variable), we tested variants that reached GWS status (p < 5 × 10−8) in the discovery phase using the following mixed model:
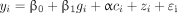 (3)
(3)This model essentially tested the main effect of g without considering a moderating effect of any environmental factors. Comparing the test results from these 2 models indicated whether the genetic association was due to the variant alone or the gene × environment effect.
In addition to the 3 principal components, age and sex were included as covariates in all analyses. The genetic relationship values between each pair of participants form a matrix. We included this genetic relationship matrix in the analyses as the variance component corresponding to the term zi in models 2 and 3 to account for the genetic relationship random effect. All association tests were performed using Gemma software,49 which allowed use of the genetic relationship matrix in the association models. We performed meta-analysis to combine association results from the discovery and replication phases using METAL.50
We tested 3 quantitative CUD-related traits in our study: the DSM-5 diagnostic criterion count and the membership likelihood scores for subtypes 4 and 5. All participants in the sample were phenotyped for these 3 traits, including those who met no or very few diagnostic criteria and would be considered to be healthy controls according to a diagnostic standard such as the DSM-IV. In contrast, only participants who were ever exposed to cocaine and passed quality control (see Genotyping and quality control, above) were included in the association analysis.
Results
Table 1 provides sample sizes by site and population group; the numbers in parentheses indicate the number of participants in the subtyping analysis. Sample characteristics are included in Table 2 (additional information about the sample has been published previously24).
Sample size by phase, site and population group
Sample characteristics
We identified 5 subtypes through cluster analysis, 4 of which included cocaine users. The most highly heritable, heavy-cocaine-use clusters were subtypes 4 and 5: narrow-sense heritability (h2) was 0.66 and 0.64, respectively; and 98.4% and 99.5% of participants met DSM-IV cocaine dependence diagnostic criteria, respectively (Appendix 1, Table S2). Subtype 4 was the largest cocaine use subgroup (n = 3258), characterized by a low rate of intravenous cocaine injection (lowest among the 4 cocaine use subtypes, Appendix 1, Table S3). Subtype 5 (n = 1916) included participants who used cocaine most heavily, were most likely to use it intravenously and had the most adverse effects from their cocaine use (e.g., 74.0% of participants reported using cocaine intravenously and 64.4% had been arrested or had trouble with the police because of cocaine use, both significantly higher than in the other subgroups). Subtype 5 also reported the earliest age of onset of both cocaine use (mean ± standard deviation [SD] 17.9 ± 4.3 yr) and the heaviest period of cocaine use (mean ± SD 25.8 ± 8.4 yr). The mean and SD of the membership likelihood for subtypes 4 and 5 among all participants are shown in Table 2. Subtype 4 had more AAs than EAs in both the discovery and replication samples (Appendix 1, Table S4). Consequently, more AAs had a high membership likelihood for this subtype than EAs. Subtype 5 was the opposite, including significantly more EAs than AAs.
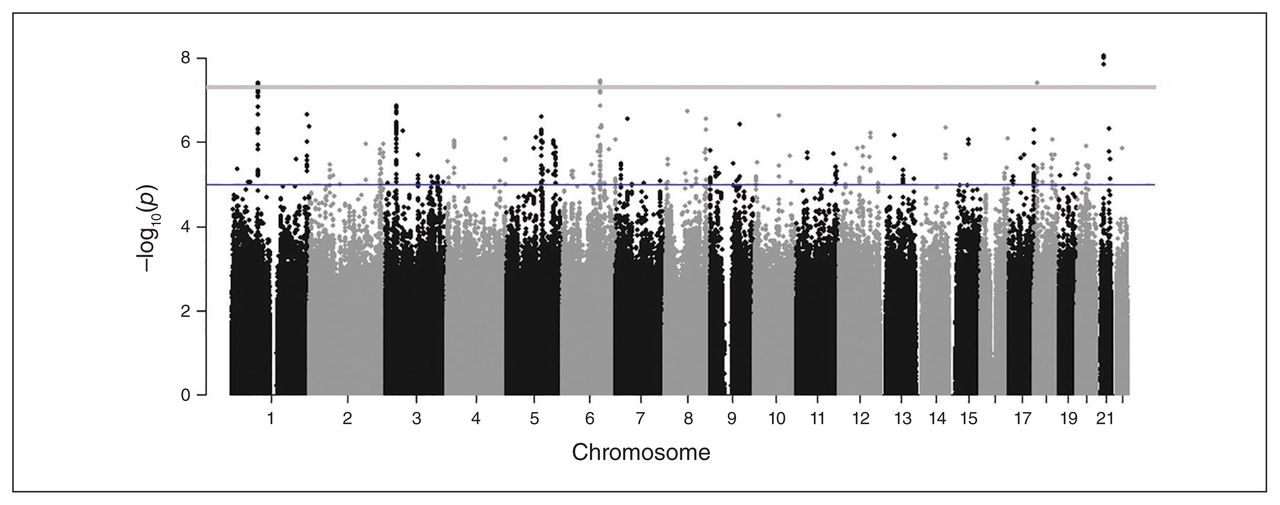
The GWAS identified a total of 24 GWS (p < 5 × E−8) loci in 13 distinct genomic regions for which the effect on CUD was moderated by environmental factors (see Fig. 1 and Appendix 1, Table S5, and Figures S3, S5, S7, S9, S11, S13 and S15), with little evidence of genomic inflation (λ = 1.002–1.076, Fig. 2; Appendix 1, Figures S4, S6, S8, S10, S12, S14 and S16). Table 3 shows the association results for the loci that were most representative of each region, evidenced by the highest imputation quality, lowest p-value or both. Of the 13 GWS loci, 11 were identified with the 2 subtypes of CUD, especially subtype 5 (the heaviest, earliest-onset subtype). In contrast, for the trait based on the DSM-5 diagnostic criterion count, only 2 variants (rs10188036 and del-13:61274071) were GWS. From these results, the most homogeneous CUD traits, subtypes 4 and 5, yielded the most novel genetic loci in association tests. Table 4 provides additional association results for the 13 GWS loci from tests that were performed separately among participants with and without exposure to the corresponding childhood environmental factors in the discovery sample.

Manhattan plot showing results from a genome-wide association study of the membership score of subtype 5 in African Americans, moderated by household tobacco use (a childhood environmental factor).

Quantile–quantile plot showing the observed distribution of p values compared with the expected distribution for the genome-wide association study of the membership score of subtype 5 in African Americans, moderated by household tobacco use (a childhood environmental factor).
Results of association tests for genome-wide significant variants in the discovery sample
Association between imputed alternative allele dosage of variants and phenotypes (Table 3), with and without exposure to the corresponding childhood environmental factors in the discovery sample*
All loci except del-1:15511771 showed associations only when the moderating effect of environmental factors was considered. For instance, the LPHN2 SNP rs149843442 was GWS for subtype 5 in AAs (p = 3.92 × E−8) only when the effect of household tobacco use was considered in the association test. For the same subtype, rs114492924 in LINC0141 was GWS in AAs only when the change in residence variable was considered. For del-1:15511771, the association test result was 2 orders of magnitude more significant (i.e., the p-value went from 2.16 × E−8 to 3.61 × E−10 when the interaction effect involving nontraditional parental care was included in the equation). Of the 13 GWS results, 11 were observed in AAs, the 2 exceptions being rs71428385 in the fibronectin 1 gene (FN1) and rs56337958 in TENM3. Both SNPs were associated with subtype 4 in EAs (Table 3) only when the interactive effect of an environmental factor was included in the model (e.g., household tobacco use for rs71428385 and traumatic experience for rs56337958). In AAs, 2 SNPs—rs10188036 in TRAK2 and del-13:61274071 in LINC00378—were also GWS for the DSM-5 criterion count under the interactive effect of change in residence and household drinking and illicit drug use, respectively. All of these results demonstrate that these loci were detectable only when considering environmental interactions in the statistical models.
Of the 13 representative loci, 7 were present in the replication data set with good imputation quality (Appendix 1, Table S5). The interaction effect of rs114492924 in LINC01411 (which encodes a non-protein-coding RNA) with change in residence was successfully replicated (p = 3.63 × E−3). Three other loci remained GWS after the results from the discovery and replication phases were combined via meta-analysis: rs10188036 in TRAK2 (meta p = 2.95 × E−8) with the presence of change in residence, rs149843442 near LPHN2 (approximately 77 000 bp from the 3′ end of the gene; meta p = 3.50 × E−8) with the presence of household tobacco use, and del-1:15511771 in TMEM51 (meta p = 9.11 × E−10) with the presence of nontraditional parental care.
Discussion
To the best of our knowledge, this is the first GWAS for CUD that considered both the phenotypic heterogeneity of the disorder and gene–environment interplay, which examined 5 informative childhood environmental factors: change in residence, nontraditional parental care, traumatic experience, household drinking and drug use, and household tobacco use. The GWAS was conducted separately for AAs (n = 2070) and EAs (n = 1570). An independent sample of AAs (n = 918) and EAs (n = 1382) was subsequently used to replicate and extend the findings through meta-analysis. Our results show that it is necessary to account for both of these issues when searching for the genetic causes of CUD. Our finding that more loci were identified for specific CUD subtypes than for the nondifferentiated general CUD trait based on diagnostic criterion count illustrates the importance of identifying clinically homogeneous CUD subtypes. In addition, 12 of the 13 representative findings were not identified in main effect tests, but could be detected only when environmental interplay was included in the statistical association models.
Our strongest finding, the one that was GWS in the discovery phase (p = 1.23 × E−8) and subsequently replicated in the independent sample (p = 3.63 × E−3), was for rs114492924 in LINC01411, which encodes a long intergenic non-protein-coding RNA. The association was evident only in the AA population when the moderating effect of a change in residence was considered. Participants with the rs114492924*T allele had higher membership likelihood for subtype 5 if they experienced a change in residence by age 13 (β = 0.27; p = 2.32 × E−8; Table 4). This association was not evident in participants who had no such childhood experience (β = 0.02; p = 0.72; Table 4). Although the biological function of LINC01411 is unknown, according to GTEx51 it is predominantly expressed in brain (Appendix 1, Figure S17), supporting its potential role in CUD risk.
Three additional variants that were GWS in the discovery phase but not in the replication phase were GWS after association results from the 2 phases were combined via meta-analysis. We found that del-1:15511771 was associated with subtype 5 when the moderating effect of nontraditional parental care was taken into account (meta p = 9.11 × E−10). The “TG” deletion was associated with a higher membership likelihood for subtype 5 only in AAs who had nontraditional parental care by age 13 (β = 0.23; p = 4.99 × E−10; Table 4). The association was not evident in those who were not exposed to this environmental factor (β = −0.05; p = 0.34; Table 4). This deletion variant is in TMEM5, which encodes a multi-pass transmembrane protein. GTEx data51 show that the gene is expressed in a wide range of human tissues, including brain (Appendix 1, Figure S18). The mechanism of this gene’s effects on CUD risk is unclear. However, previous data show that the transmembrane protein coded by this gene interacts with many chemicals, such as phenobarbital and benzopyrene.52 Thus, it would be of interest to investigate how this protein interacts with cocaine.
Another variant that, on meta-analysis, was GWS for subtype 5 was rs149843442 (meta p = 3.50 × E−8), but was GWS only when the moderating effect of household tobacco use was considered. The A allele of this SNP was associated with a higher membership likelihood for subtype 5 in AAs who experienced household tobacco use by age 13 (β = 0.23; p = 1.04 × E−7; Table 4), an effect that was not evident in participants who were not exposed to this environmental factor (β = −0.05; p = 0.50; Table 4). HaploReg53 shows that rs149843442 alters 5 regulatory motifs (Appendix 1, Table S7) and is in a genomic region that overlaps with 2 potential regulatory elements indicated by chromatin modification H3K4me1 (Appendix 1, Table S6). Thus, this SNP could be functional. On the other hand, rs149843442 is in perfect linkage disequilibrium with rs6685582 (r2 = 1, estimated using the African population in the 1000 Genomes Project54), which was also GWS for subtype 5 in the discovery phase (p = 4.62 × E−8), which was also moderated by household tobacco use. However, no data were available in the replication sample to replicate this finding. We know that rs6685582 is in a very active genomic region that overlaps with dozens of regulatory elements identified in various human tissues, including brain (Appendix 1, Table S8), and it alters 2 regulatory motifs (Appendix 1, Table S9). Therefore, the association involving rs149843442 could also be positional, tagging rs6685582 or other variants that are in close linkage disequilibrium. The regulatory elements overlapping both variants are likely involved in the regulation of LPHN2, which is their closest gene. The 2 variants are ~77 000 to 96 000 bp from the 3′ end of the gene. LPHN2 encodes a member of the latrophilin subfamily of G-protein-coupled receptors and participates in the regulation of exocytosis. This gene has been linked to several human disease phenotypes,55–59 including 2 related to brain function: electroencephalogram60 and target recognition in entorhinal–hippocampal synapse assembly.61
The third GWS finding emerging from meta-analysis was for rs10188036, associated with the DSM-5 diagnostic criterion count in interaction with the environmental variable change in residence. The rs10188036*C allele was associated with a lower DSM-5 diagnostic criterion count only in AAs who experienced change in residence by age 13 (β = −1.91, p = 1.20 × E−8, Table 4, versus β = 0.96, p = 0.16, for those who had not moved). This SNP is in the trafficking kinesin-binding protein 2 gene (TRAK2) on chromosome 2. The TRAK2 protein appears to regulate endosome-to-lysosome trafficking of membrane cargo and has been linked to cholesterol efflux and HDL biogenesis,62 as well as late-onset Alzheimer disease.63 More relevant to the current study, TRAK2 interacts with the γ-aminobutyric acid A (GABAA) receptor. Cocaine potentiates GABA release and leads to the inhibition of dopamine neurons, thus driving drug-adaptive behaviour.64 Therefore, TRAK2 could affect the susceptibility of CUD through its protein product’s effect on the GABAA receptor.
We also identified 7 loci that had a GWS association with CUD (5 in AAs and 2 in EAs in the discovery phase) but that could not be tested for replication due to the lack of available data (Table 3). The most notable of these SNPs was rs148009780 in the synaptogyrin (SYNGR1) gene on chromosome 22. The rs148009780*T allele was associated with higher membership likelihood for subtype 5 in AAs who experienced a change in residence (β = 0.24, p = 9.14 × E−8, Table 4, versus β = −0.04, p = 0.56, for those who did not). SYNGR1 encodes an integral membrane protein associated with pre-synaptic vesicles in neuronal cells and is most highly expressed in brain (Appendix 1, Figure S19). Thus, it is a biological candidate for disorders related to the central nervous system and variation in the gene has been associated with schizophrenia and bipolar disorder in a southern Indian population65 and schizophrenia in an Italian sample.66 Moreover, a recent study identified a genomic region near SYNGR1 that is in close linkage to alcohol dependence (LOD = 3.2) in an AA sample.67 Our findings suggest that SYNGR1 may also regulate susceptibility to CUD. Another variant that may be worth further investigation is rs56337958, an intronic SNP in TENM3 on chromosome 4. The rs56337958*G allele was associated with higher membership likelihood for subtype 4 in EAs who had a traumatic experience by age 13 (β = 0.31, p = 4.29 × E−8, Table 4, versus β = 0.02, p = 0.64, for those who did not). According to GTEx data,51 TENM3 encodes a large transmembrane protein that may be involved in the regulation of neuronal development and is expressed in many brain tissues (Appendix 1, Figure S20).
Our previous study showed the FAM53B SNP rs2629540 to be associated to CUD criterion count in AAs.9 However, we did not identify any variants in or near FAM53B that were GWS in the current study, possibly because the phenotypic definitions and covariates in this study differed from those in the previous study. Here, the strongest signal for an association of rs2629540 with CUD was found with subtype 4 among AAs when taking into account the moderating effect of a change in residence (p = 1.01 × E−4). There are 2 possible explanations for the weaker support for the association of FAM53B with CUD. First, the GWAS sample was smaller in present study (3640 total) than in the previous study (5697 total),9 because we excluded people with no previous cocaine exposure or no information on environmental factors. Second, as noted above, the CUD-related traits tested differed in the 2 studies. In the previous study, we used 2 binary traits, cocaine dependence and cocaine-induced paranoia, and 1 quantitative trait, the DSM-IV criterion count, which contrasted with the subtypes and DSM-5 criterion count used here.
Although childhood traumatic experience has been shown to have a profound impact on adulthood substance use,38,39 fewer genetic variants were associated with CUD when accounting for its effect compared with the effects of other environmental factors. A possible explanation for this finding is that, compared with other factors, fewer participants in our study had had a traumatic experience by age 13, substantially limiting the power to detect associated variants.
Limitations
The findings in this paper should be viewed in the context of a number of limitations. The main limitation is the lack of availability of clinical data and high-quality genotypes to replicate 6 of the 13 loci that were GWS in the discovery phase. Despite the fact that databases such as dbGap (www.ncbi.nlm.nih.gov/gap) include genotyped participants with CUD, the phenotypic variables used in our cluster analysis and the environmental variables used in the association study were specific to the SSADDA diagnostic interview and were not available in dbGap data sets. Variation in measurements among studies is a well-recognized problem in data aggregation.17 The 3 non-replicated loci and the 6 loci without data for replication could represent false-positive findings from GWAS, especially those for which there was no previous evidence supporting their potential involvement in the biological processes contributing to CUD. Nonetheless, among the 7 loci for which replication data were available, 4 were GWS either in replication or after combining results from the 2 analytic phases through meta-analysis, so they are worthy of further investigation. Another limitation was that the method used to derive the 5 binary environmental factors may not have been optimal. We assigned a value of 0 or 1 to participants using a threshold based on the distribution of responses to ensure an adequate number of participants who were exposed to the environmental effects. In addition, correction may have been needed for multiple statistical testing. Because the subtype quantitative traits were defined by classifying participants in one cluster from those outside the cluster, these traits were expected to be correlated. Because there was correlation among the traits (Appendix 1, Figure S21) and among the environmental factors (Appendix 1, Figure S22), and the hypotheses for EAs and AAs were distinct, Bonferroni correction was too restrictive. Adjustment methods may need to be developed to appropriately correct for testing multiple correlated traits and environmental factors in 2 populations. Moreover, the overlap in genotyped markers between the discovery and replication samples was relatively small due to the different genotyping microarrays. However, an advantage of using the samples was that the participants in the discovery and replication samples were identically assessed by a well-validated procedure, resulting in high confidence and consistency in the phenotypes and environmental factors. Lastly, most of the variants that were identified with association were imputed, but all had excellent INFO scores for the imputation (Appendix 1, Table S1).
Conclusion
We designed a genome-wide approach to detect gene–environment interactions that could be associated with more refined disease phenotypes. We identified a locus, rs114492924, that was associated with CUD in AAs. The SNP reached GWS in the discovery sample and was replicated in a separate sample. Three additional loci reached GWS in AAs when the discovery and replication samples underwent meta-analysis, and 9 other loci were GWS in the discovery sample only. Although replication is required to validate our findings, many of the identified loci have collateral support from other sources, such as gene expression studies and the localization of transcriptional regulatory elements and motifs, supporting their relevance to the risk of CUD. Based on the findings in this study, models used to test samples collected for identifying genetic variants that contribute to psychiatric traits such as CUD should include psychometrically established measures of environmental effects and account for the phenotypic heterogeneity of the trait.
Footnotes
Funding: This work was supported by NIH grant R01DA037349 and NSF grants DBI-1356655 and CCF-1514357. J. Bi was also supported by NIH grant K02DA043063.
Competing interests: H. Kranzler is a member of the American Society of Clinical Psychopharmacology Alcohol Clinical Trials Initiative, which was supported in the last 3 years by AbbVie, Alkermes, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, Pfizer, Arbor, and Amygdala Neurosciences. H. Kranzler and J. Gelernter are named as inventors on PCT patent application #15/878,640 entitled, “Genotype-guided dosing of opioid agonists,” filed January 24, 2018. J. Sun and J. Bi declare no competing interests.
Contributors: J. Sun and J. Bi designed the study. H. Kranzler and J. Gelernter acquired the data, which J. Sun and J. Bi analyzed. J. Sun and J. Bi wrote the article, which all authors reviewed. All authors approved the final version to be published and can certify that no other individuals not listed as authors have made substantial contributions to the paper.
- Received June 15, 2018.
- Revision received November 29, 2018.
- Revision received February 13, 2019.
- Accepted April 6, 2019.
References
In this issue

{kind=link}
{kind=link}
Article tools


