

Abstract
The selective serotonin reuptake inhibitors (SSRIs) are the most frequently prescribed antidepressant drugs, because they are well tolerated and have no severe side effects. They rapidly block serotonin (5-HT) reuptake, yet the onset of their therapeutic action requires weeks of treatment. This delay is the result of presynaptic and postsynaptic adaptive mechanisms secondary to reuptake inhibition. The prevention of a negative feedback mechanism operating at the 5-HT autoreceptor level enhances the neurochemical and clinical effects of SSRIs. The blockade of 5-HT2A receptors also seems to improve the clinical effects of SSRIs. These receptors are located postsynaptically to 5-HT axons, mainly in the neocortex. Pyramidal neurons in the prefrontal cortex are particularly enriched in 5-HT2A receptors. Their blockade may affect the function of prefrontal–subcortical circuits, an effect that probably underlies the beneficial effects of the addition of atypical antipsychotic drugs, which are 5-HT2A receptor antagonists, to SSRIs in treatment-resistant patients.
Introduction
The World Health Organization estimates that unipolar depression will be the second most prevalent cause of illness-induced disability by the year 2020.1 Not surprisingly, antidepressants are the third most commonly sold group of therapeutic agents worldwide. Most of these treatments are based on molecules that target a single protein in the brain, the serotonin (5-HT) transporter. These agents, the selective serotonin reuptake inhibitors (SSRIs), which inhibit 5-HT reuptake, account for about 80% of all antidepressants on the market. Other antidepressant drugs such as the serotonin and noradrenaline reuptake inhibitors (SNRIs) or the classic tricyclic antidepressants (e.g., amitryptyline, clomipramine, imipramine) inhibit the reuptake of noradrenaline as well. Some of these old drugs, such as clomipramine, have a complex pharmacology and have been proved to be the best antidepressant treatments for severe depression,2,3 although the presence of their many severe side effects is a very serious limitation to their use. Indeed, the success of the SSRIs lies mainly in their safety, better tolerability and absence of severe side effects compared with the tricyclic drugs, which improves compliance and quality of life of the patient with depression.
Intensive research efforts have led to the identification of many pharmacologic effects of antidepressant drugs. However, this knowledge has not been efficiently translated into new and more rapid and effective medicines. Indeed, it should be noted that most antidepressant drugs act indirectly, that is, by enhancing the 5-HT tone on 1 or more 5-HT receptors through the inhibition of reuptake (or deamination in the case of monoamine oxidase inhibitors [MAOIs]). A few antidepressant drugs (nefazodone, trazodone, mirtazapine) are antagonists of certain receptors, such as 5-HT2A or α2-adrenoceptors, a property that may underlie their therapeutic properties. Perhaps the 5-HT receptor more directly linked with the antidepressant effects of SSRIs has been the 5-HT1A receptor. On the one hand, preclinical studies have shown an increase of 5-HT1A receptor-mediated hippocampal transmission after long-term treatment with SSRIs and other antidepressant drug classes.4 Despite this experimental evidence, for various reasons, most selective 5-HT1A agonists developed so far have failed to demonstrate clinical effectiveness. Indeed, the clinical effectiveness and use of the only marketed compound of this class (buspirone) is very far from that of other antidepressants, despite claims in favour of the use of 5-HT1A agonists.5 On the other hand, presynaptic 5-HT1A autoreceptors are a primary target of several types of antidepressant drug that enhance extracellular 5-HT (SSRIs, MAOIs) or act directly on such receptors.
Ideally, new antidepressant drugs should be targeted at the postsynaptic receptor(s) or intracellular signalling pathways responsible for the therapeutic effects of existing drugs. In this way, they would overcome the neuronal adaptive mechanisms (both presynaptic and postsynaptic) that delay and limit their therapeutic action. However, the identification of these mechanisms is hampered by many factors, such as the intrinsic complexity of the study of brain function, difficulties in assessing the effects of antidepressant drugs in humans and the lack of reliable animal models of depression. To a large extent, the development of antidepressant drugs has been serendipitous or empirical. Only in a few instances have new therapeutic strategies been based on neurobiologic grounds.
Possibly because of this indirect action, current antidepressants pose 2 main problems: less than optimal effectiveness and slow onset of action. Hence, they exert their initial pharmacologic action in hours, but they require prolonged administration before significant clinical improvement occurs. Typically, the response rate for SSRIs is 60% at 6 weeks, where response is defined as a 50% reduction of the initial severity. If one considers remission, rates drop to 35%–40% at 6 weeks. In a high proportion of patients, treatment must proceed for years to prevent relapses and recurrences adequately. The initial delay in clinical action results from neurobiologic adaptive mechanisms secondary to the activation of the initial pharmacologic target. These encompass presynaptic changes in the activity of monoamine-containing neurons and postsynaptic changes in corticolimbic areas, possibly involving changes in gene expression, that reshape the function of brain circuits altered in major depression.6 Hence, it is clear that we are still far from the ideal antidepressant, that is, one with a well-defined, direct target (a postsynaptic receptor or intracellular messenger) and high effectiveness and rapid (< 1 wk) onset of action.
The 5-HT system
An extensive review of the characteristics of the serotonergic system is beyond the scope of the present article. The reader is referred to several review papers dealing with the anatomy, physiology, neurochemistry and neuropharmacology of this neurotransmitter.7–10 However, we would like to underline a few characteristics of 5-HT neurons that are deemed important for a better understanding of the neurobiologic effects of antidepressant drugs.
First, there are few 5-HT neurons whose cell bodies are concentrated in the raphe nuclei of the midbrain. For example, it has been estimated that the human brain contains about 250 000 5-HT neurons of a total of 1011 neurons.7 Second, 5-HT neurons are extensively arborized, and their axons reach all brain areas. For instance, the rat hippocampus contains a density of 1–4 × 106 serotonergic varicosities/mm3.11 They hardly make synaptic contacts, because they release 5-HT in a paracrine manner.7,12 Third, 5-HT neurons are tonically active with a slow and regular pacemaker-type activity that ceases during rapid eye movement sleep.7 These 3 characteristics, in combination, make changes in the firing activity of 5-HT neurons extremely important for the overall function of the 5-HT system, because they will influence in a concerted manner a large population of target neurons in the forebrain.
The activity of 5-HT neurons is tightly controlled by a number of afferent pathways, mainly including glutamatergic inputs from forebrain areas such as the prefrontal cortex (PFC), a tonic noradrenergic input from various pontine nuclei and inhibitory γ-aminobutyric acid (GABA)-ergic inputs from local interneurons.10 The role of other transmitters such as histamine or acetylcholine and peptides (e.g., substance P, corticotropin-releasing factor, cholecystokinin, hypocretin-orexin) is still poorly understood, yet new information is emerging. Finally, a very important mechanism of control of 5-HT neurons is self-inhibition through 5-HT1A autoreceptors. Activation of these receptors by 5-HT leads to opening of potassium channels in the cell membrane, hyperpolarization of the cell and a cessation of cell firing. 13,14 Local release of 5-HT in the raphe nuclei from axonal collaterals or crosstalk between different 5-HT neurons will thus diminish neuronal firing and produce a negative feedback regulation of transmitter release.15,16 Selective 5-HT1A receptor agonists also elicit the same effect by interacting with raphe 5-HT1A receptors.17,18 In addition, 5-HT1B/1D receptors located on nerve terminals respond to 5-HT released locally in the terminal fields to inhibit further transmitter release.19 These 2 mechanisms ensure tight feedback control of the activity of serotonergic neurons and of terminal 5-HT release.
Antidepressants and 5-HT1A autoreceptor desensitization
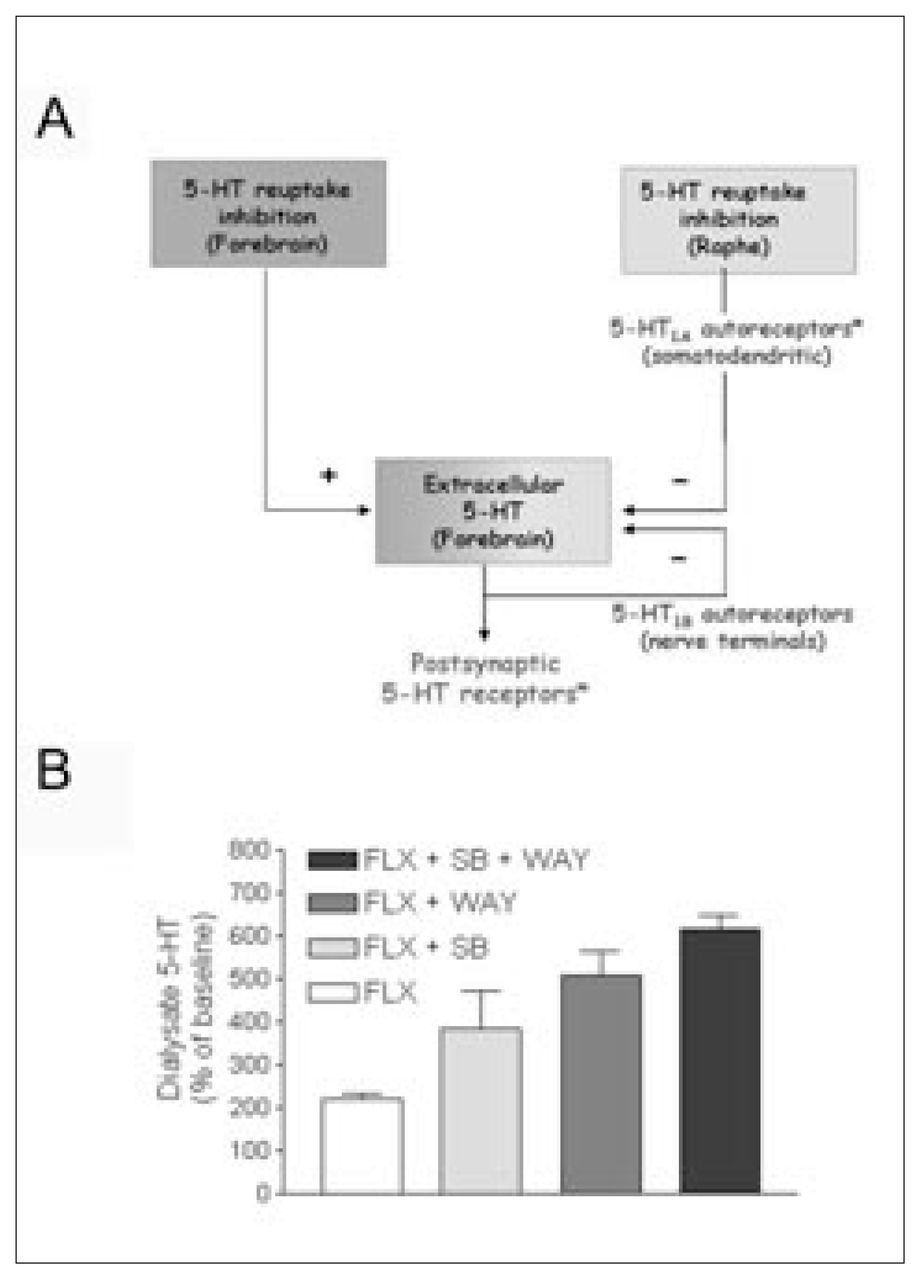
5-HT1A receptors are deeply involved in the mechanism of action of antidepressant drugs. They occur in mammalian brain in 2 different populations: on 5-HT neurons of the midbrain raphe nuclei (autoreceptors) and on neurons postsynaptic to 5-HT nerve terminals, mainly in cortico-limbic areas. In both regions, 5-HT1A receptors have a somatodendritic location. The activation of 5-HT1A receptors increases potassium conductance, thus hyperpolarizing the neuronal membrane and reducing the firing rate of serotonergic and pyramidal neurons in the cortex and hippocampus.13,20–22 Most antidepressant drugs increase the concentration of 5-HT in the extracellular brain space by preventing its reuptake. However, this increase is offset by a negative feedback operating at the 5-HT cell-body level (Fig. 1). Using the technique of in-vivo microdialysis, it was shown that the inhibition of 5-HT reuptake produced by single administration of the tricyclic antidepressant clomipramine and the SSRIs caused a marked enhancement of the extracellular concentration of 5-HT in the midbrain raphe nuclei.15,23,24 This effect was greater than in the forebrain25,26 and accounted for the suppression of 5-HT cell firing induced by various antidepressant drugs that block 5-HT reuptake.8,27 The 5-HT1A autoreceptor-mediated inhibition of cell firing was accompanied by a reduction of terminal 5-HT release, which thus attenuated the increase in extracellular 5-HT produced by reuptake blockade.15,16,24 Consequently, the activation of postsynaptic 5-HT receptors responsible for the therapeutic effect is lower than expected. Terminal autoreceptors further limit the increase in synaptic (extracellular) 5-HT produced by SSRIs in different species.28,29
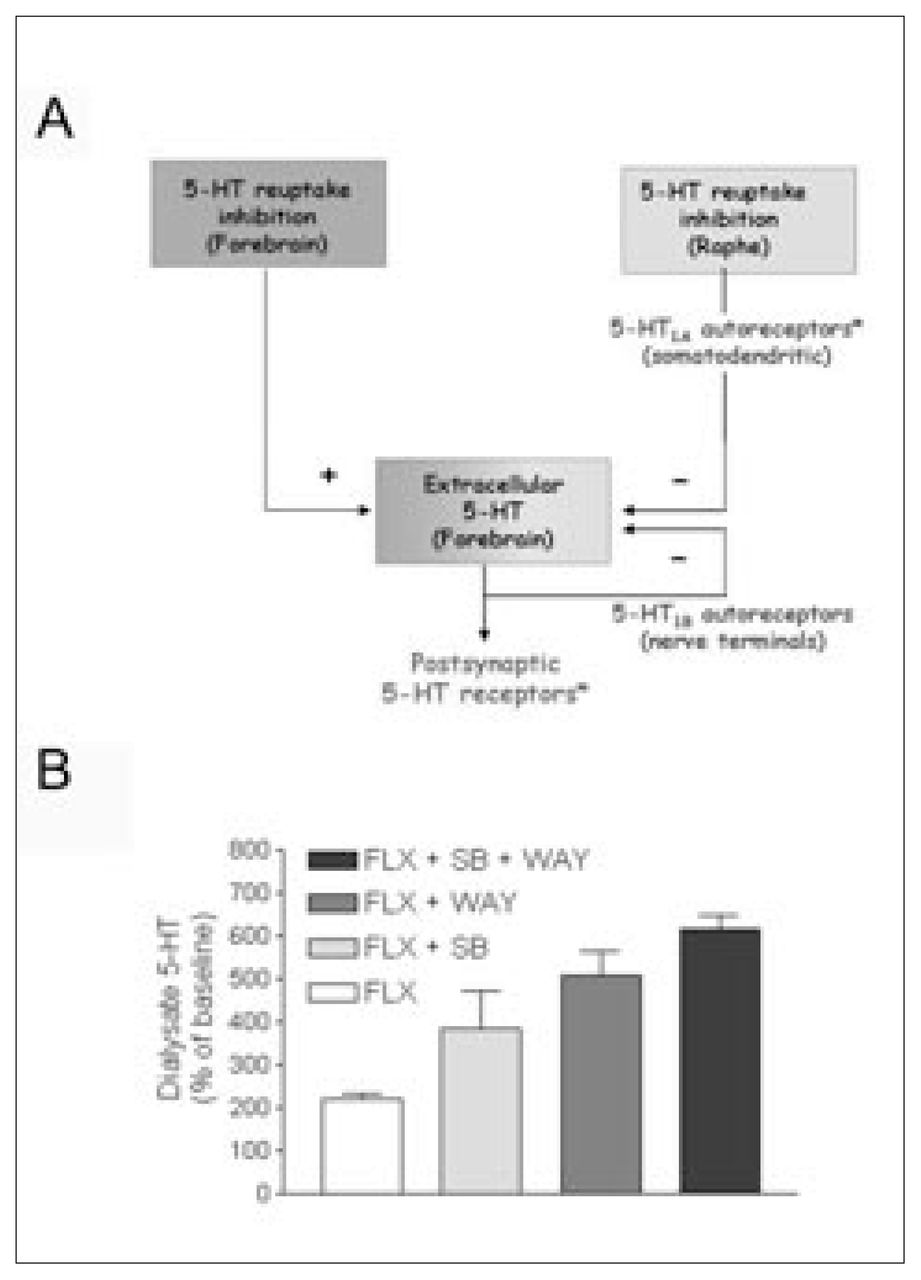
Inhibition of serotonin (5-HT) reuptake in the forebrain by selective serotonin reuptake inhibitors (SSRIs) increases extracellular 5-HT. This effect is attenuated by the reduction in 5-HT release that follows the activation of 5-HT autoreceptors by the SSRI-induced excess in 5-HT. The increase in extracellular 5-HT is particularly remarkable in the midbrain raphe nuclei, which contain the cell bodies of 5-HT neurons. 5-HT1A receptors are then activated by 5-HT, released from cell bodies and dendrites and from axons within the raphe nuclei, which causes an inhibition of cell firing and, subsequently, of impulse-dependent terminal 5-HT release. The activation of terminal (5-HT1B) autoreceptors also reduces 5-HT release. Asterisks denote the possible sites of action of pindolol in the human brain (unlike in rodents, pindolol lacks significant affinity for human 5-HT1B receptors).
B: Autoreceptor antagonists potentiate the effects of SSRIs. Microdialysis experiments in rats indicate that the blockade of 5-HT1A and/or 5-HT1B receptors with selective antagonists (WAY100635, 0.3 mg/kg subcutaneously, and SB224289, 4 mg/kg intraperitoneally, respectively) potentiates the effects of the administration of the SSRI fluoxetine (FLX) (10 mg/kg intraperitoneally) on extracellular 5-HT in the frontal cortex. Results are mean values (and standard error of the mean) of extracellular 5-HT. Permission to publish this modified figure was received from Elsevier (Trends Pharmacol Sci 2001;22:224–8).33
However, the efficacy of this negative feedback resulting in attenuation of cell firing and terminal 5-HT release is less marked after long-term treatment with SSRIs. Thus, long-term SSRI treatment resulted in a recovery of the firing of 5-HT cells in the dorsal raphe nucleus (DR) and an increase in extracellular 5-HT greater than after single administration.8,30 Both effects are likely to result from the 5-HT-induced desensitization of raphe 5-HT1A autoreceptors.8
Potential use of 5-HT1A autoreceptor blockade: the case of pindolol
In 1993, one of us (F.A.) proposed that “5-HT1A receptor antagonists could accelerate (and perhaps augment) the clinical effects of antidepressants by preventing this negative feedback.”31 This would enable a more rapid increase of synaptic 5-HT, preventing the inhibition of 5-HT release observed in microdialysis studies and mimicking the 5-HT1A receptor desensitization produced by the prolonged administration of antidepressants. 8 Given the lack of selective 5-HT1A receptor antagonists for human use, this hypothesis was tested with the β-adrenoceptor/5-HT1A receptor antagonist (±)pindolol. This compound, with an affinity for 5-HT1A receptors of about 10–8 mol/L, antagonized several actions mediated by the activation of central 5-HT1A receptors, such as hypothermia or hormonal secretion. Since the first study, published in 1994,32 the results of 15 placebo-controlled clinical trials and several open-label studies using pindolol have been reported.33 In general, the addition of pindolol (7.5 mg/d) to SSRIs accelerates the antidepressant response. It is remarkable that despite current difficulties in evaluating the onset of antidepressant action and sometimes in discriminating between active drug and placebo in clinical trials, significant differences were noted in 5 of 7 trials, with a partial success in another trial. In 2 of these trials, the addition of pindolol also increased the end-point response rate of the SSRI used (from 59% to 75% with fluoxetine and from 48% to 81% with paroxetine33). Three studies also examined the ability of pindolol to improve the clinical response to SSRIs in anxiety disorders. Pindolol has been also used in treatment-resistant patients with dissimilar results. A few studies reported some benefit of the addition of pindolol, whereas others have not reported any difference versus placebo. However, an epidemiologic study reported a significantly lower incidence of depression and lower consumption of antidepressants (3-year follow-up) in patients treated with pindolol for cardiovascular purposes, compared with other β-blockers,34 which suggests an overall beneficial effect of pindolol in affective disorders.
One crucial question regarding the mechanism of action of pindolol is the occupation of central 5-HT1A receptors at the dose used (typically 7.5 mg/d). The comparison of the plasma levels of pindolol in treated patients (about 25 nmol/L)35 with the in-vitro affinity of pindolol for human 5-HT1A receptors obtained in autoradiographic studies36,37 suggests that this pindolol dose occupies 5-HT1A receptors in the human brain. This view has been confirmed in positron emission tomography (PET) studies. In one of these, pindolol administration (7.5 mg/d for 1 wk) to healthy volunteers produced a significant decrease in [11C]WAY100635 binding and higher occupancy in the DR (40%) than in the hippocampus (18%).38 Another PET scan study yielded lower occupancy results in both areas and a presynaptic versus a postsynaptic difference in receptor occupancy39 in agreement with animal data that support a preferential action of pindolol on somatodendritic 5-HT1A receptors.40,41 One of the conclusions of these studies is that higher dosages (e.g., 3 × 5 mg/d or greater) should be tested in future augmentation trials to increase the occupancy of 5-HT1A autoreceptors.
Because selective 5-HT1A receptor antagonists augment the neurochemical and behavioural effects of SSRIs, such agents, devoid of the β-blocking properties of pindolol, should be tested in clinical trials to determine whether blockade of 5-HT1A receptors may augment the clinical effects of SSRIs. An important concern is the lack of selectivity of these new agents for presynaptic versus postsynaptic 5-HT1A receptors.42 The full blockade of postsynaptic receptors may cancel the increased transmission through hippocampal 5-HT1A receptors produced by antidepressant drugs in the rat brain.4 However, because other 5-HT receptors may also be involved in the effects of SSRIs, the hypothesis needs to be experimentally tested in clinical trials.
5-HT2A receptors: a possible role in the augmentation of antidepressant response
In recent years, a number of open-label and placebo-controlled studies have suggested that atypical antipsychotic drugs and some antidepressants (e.g., mirtazapine and mianserin) augment the clinical response to SSRIs in treatment-resistant patients.43–46 One common feature of these agents is their ability to occupy 5-HT2 receptors in the brain at clinical doses and to block 5-HT2-mediated responses, in particular those mediated by 5-HT2A receptors.47 Likewise, many antidepressants downregulate 5-HT2A receptors after repeated treatment.48 Altogether, these observations support a role for 5-HT2A receptors in antidepressant drug action. These receptors are mainly localized in the neocortex, and its selective blockade by M100907 augments the antidepressant effect of SSRIs in the differential reinforcement of low rate 72 seconds (DRL-72 s) schedule, a task related to PFC function. This effect does not involve a presynaptic potentiation of the increase in 5-HT produced by the SSRI, which suggests that the improvement in executive functions arises from the blockade of postsynaptic 5-HT2A receptors.49
PFC, major depression and the 5-HT system
The hippocampus has been the focus of many studies of and theories about the pathophysiology and treatment of depression. More recent views on this issue emphasize the role of the plastic changes in this subcortical brain structure.6 However, the PFC also plays a major role in depression. Hence, brain imaging studies have consistently shown an association between major depression and hypoactivity of the prefrontal lobe.50,51 Furthermore, stroke in the left PFC is associated with a high incidence of major depression.52 These observations suggest that the PFC plays a main role in depression.
Because of its unique cytoarchitecture and connectivity, the PFC is deeply involved in higher brain functions and exerts a top–down control of brain functions through the processing and integration of signals from other brain areas, including large parts of the neocortex, some thalamic nuclei and the brain stem.53,54 Signal integration in pyramidal neurons is exerted at various cellular levels, including apical and basal dendrites, cell bodies and the axon hillock. The apical dendrites are highly enriched in serotonergic 5-HT2A receptors, which are also present in large and medium-sized GABAergic interneurons that control the activity of pyramidal neurons in local microcircuits.55–59 Compounds such as lysergic acid diethylamide (LSD) or 2,5-dimethoxy-4-iodoamphetamine (DOI) likely exert their hallucinogenic action through a massive activation of 5-HT2A receptors, whereas atypical antipsychotic drugs are antagonists at 5-HT2A receptors.60,61
Interestingly, in the rodent brain, the medial PFC (mPFC) innervates, via long glutamatergic axons, a number of subcortical brain areas that are potentially involved in depressive symptomatology such as the nucleus accumbens (anhedonia), the amygdaloid complex (fear, anxiety), limbic structures (depressed mood, memory impairment), other parts of the PFC (cognitive disturbances, behavioural withdrawal) or the hypothalamus (hypothalamic–pituitary–adrenal axis, appetite, sleep, sexual drive).62 Therefore, a change in the activity of prefrontal projection (pyramidal) neurons in depression may have a strong impact on the function of these brain structures.
Moreover, there is a reciprocal connectivity between the mPFC and the brainstem aminergic nuclei. The ventral tegmental area/substantia nigra pars compacta, the raphe nuclei and the locus coeruleus give rise to the dopaminergic, serotonergic and noradrenergic innervation of most forebrain structures, including those that are potentially involved in depression (with the exception of the substantia nigra pars compacta, which is more involved in motor function). The pyramidal neurons in intermediate–deep layers of the PFC play a fundamental role in prefrontal function. Owing to their large apical dendrites, they integrate incoming excitatory signals from various cortical layers and from subcortical areas (mainly the mediodorsal thalamus) and project, via long axons, to the aforementioned areas. Modulatory inputs arise from the brainstem aminergic nuclei, thus closing mPFC-brainstem circuits, whereas local control is exerted by GABAergic interneurons.62 Pyramidal neurons and GABAergic interneurons are enriched in aminergic receptors, such as 5-HT1A, 5-HT2A, 5-HT2C, 5-HT3, dopamine D1, D2 and α-adrenoceptors.
Consistent with these anatomical relationships, evidence in recent years indicates that the mPFC has a profound influence on the activity of brainstem aminergic neurons. The stimulation of this cortical area increases burst firing of dopaminergic neurons of the ventral tegmental area, and prefrontal lesions reduce the number of spontaneously active dopamine neurons. 63,64 Likewise, an excitatory input of the mPFC on noradrenergic neurons of the locus coeruleus has been documented.65 Finally, more recent observations support the notion that most 5-HT neurons of the DR are under prefrontal control.59,66,67 Given that most pharmacologic treatments of a large number of neuropsychiatric disorders target 5-HT neurons, the study of the mPFC–raphe circuit may provide new clues to understanding the pathophysiology of these disorders, including depression and schizophrenia.
Modulation of the activity of 5-HT neurons by the mPFC
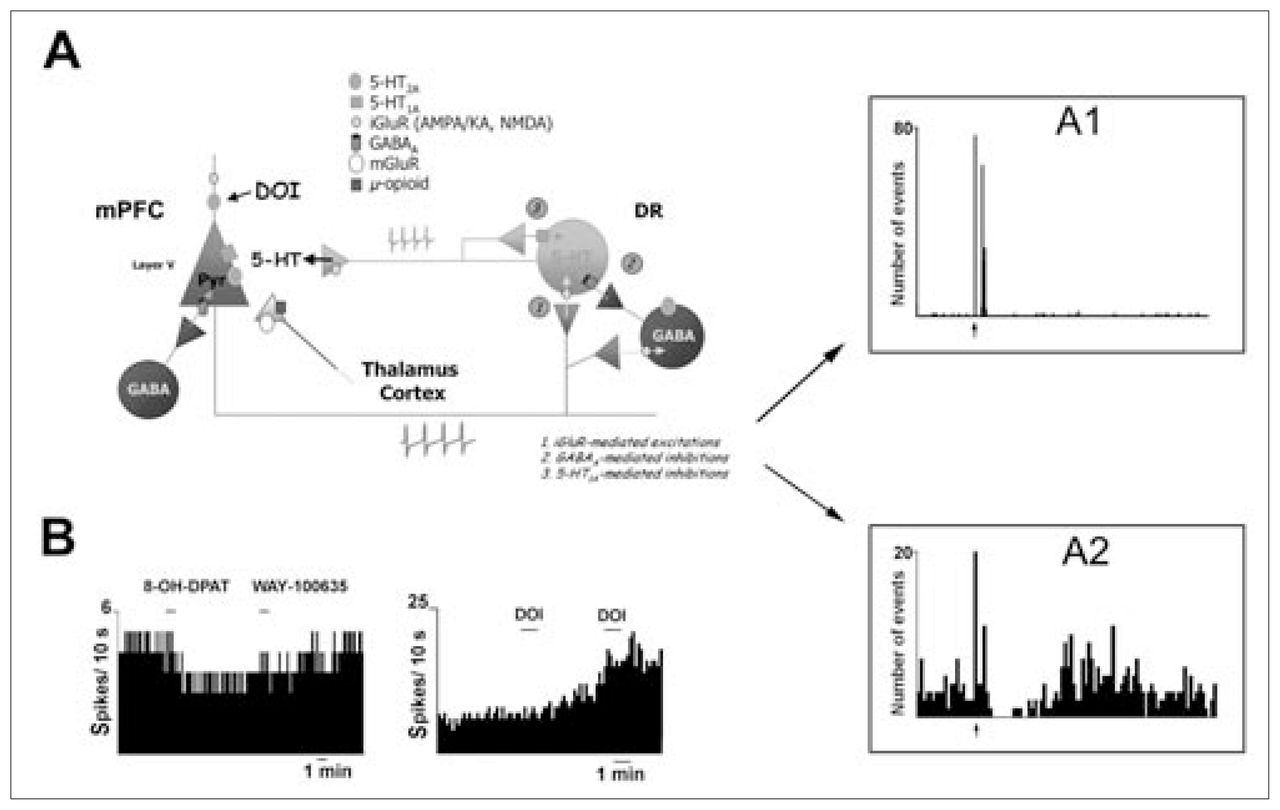
Tracing studies indicate that the mPFC innervates DR 5-HT neurons.62,66,68–71 The electrical stimulation of the mPFC at a physiologic frequency results in a short-latency (about 17 ms) monosynaptic activation of 5-HT neurons in the DR, which is mediated by ionotropic glutamate receptors 2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionate (AMPA)/kainate (KA) and N-methyl-d-aspartate (NMDA) (Fig. 2).67 Likewise, long-latency (about 35 ms) and long-duration (up to about 150 ms) inhibitions are also observed that result from 5-HT1A autoreceptor activation by recurrent collaterals or crosstalk between 5-HT neurons in response to the stimulus-triggered excitation and release of 5-HT. Likewise, descending excitatory axons from the mPFC can also inhibit 5-HT neurons via GABAergic interneurons. 67,72 Thus, the activity of individual 5-HT neurons can be finely tuned by the mPFC through direct (excitatory) or indirect (inhibitory, via 5-HT1A or GABAA receptor activation) inputs, although the overall influence of the mPFC on the bulk of 5-HT neurons appears to be excitatory. Thus, the application of the 5-HT1A and 5-HT2A agonists in the mPFC to inhibit and excite, respectively, local projection neurons resulted in a parallel modulation of the firing rate of 5-HT neurons in the midbrain.59,67 Moreover, these changes were accompanied by a similar change in 5-HT release in the mPFC,59,67 which supports the idea that postsynaptic 5-HT1A and 5-HT2A receptors in the PFC can contribute to the distal feedback control of 5-HT neuronal activity and terminal 5-HT release through the modulation of descending excitatory afferents (Fig. 2).

Medial prefrontal cortex (mPFC)–dorsal raphe (DR) circuit. Stimulation of projection neurons in the mPFC increases propagation of action potentials through descending excitatory axons that innervate the DR, among other subcortical areas. These excitatory afferents control the activity of 5-HT neurons through 3 different mechanisms: (1) directly, via N-methyl-d-aspartate (NMDA) and 2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionate/kainate (AMPA/KA) receptors; (2) indirectly, via γ-aminobutyric acid (GABA) interneurons and activation of GABAA receptors; and (3) via activation of 5-HT1A autoreceptors by recurrent collaterals or crosstalk between different 5-HT neurons. The panels on the right show peristimulus-time histograms of mPFC-induced excitation (A1, corresponding to a iGluR-mediated response; mean latency 16 ms, mean duration 17 ms) and inhibition (A2, corresponding to GABAA and/or 5-HT1A-mediated responses; mean latency 36 ms, mean duration 150 ms) recorded in DR 5-HT cells.
B: The local application in the mPFC of 8-OH-DPAT and 2,5-dimethoxy-4-iodoamphetamine (DOI), 5-HT1A and 5-HT2 receptor agonists, respectively, decreases and increases the firing rate of 5-HT neurons in the DR as a result of the inhibition and stimulation of the activity of mPFC pyramidal neurons projecting to the DR. iGluR = ionotropic glutamate receptor, mGluR = metabotropic glutamate receptor. Permission to publish these modified figures was received from the Society for Neuroscience (J Neurosci 2001;21:9856–66, J Neurosci 2001;21:9917–29).59,67
Modulation of the activity of cortical pyramidal neurons in the mPFC by 5-HT1A and 5-HT2A receptors
The PFC of the rodent, primate and human brain is densely innervated by 5-HT axons and is highly enriched in various receptors, notably the 5-HT1A and 5-HT2A subtypes.38,73–78 Clues about a role of 5-HT receptors in prefrontal function are numerous, including:
the hallucinogenic action of 5-HT2A receptor agonists (e.g., LSD, DOI) and the 5-HT2A antagonist action of atypical antipsychotic drugs, some of which are also 5-HT1A receptor agonists60,61
the involvement of prefrontal 5-HT2A receptors in working memory79
the involvement of 5-HT1A receptors in memory and anxiety80–82
the existence of 5-HT receptor abnormalities in the frontal lobe of psychiatric patients83–85
Furthermore, 5-HT1A and 5-HT2A receptors mediate the changes in cortical dopaminergic transmission induced by atypical antipsychotic drugs.86 Together, these observations support an important role for 5-HT1A and 5-HT2A receptors in the normal and pathologic function of the PFC.
Both receptors are abundantly expressed by pyramidal neurons.55–58,87–89 5-HT2A receptors are also present in GABA interneurons55,57 and catecholaminergic axons in the mPFC.89 The activation of 5-HT1A receptors hyperpolarizes prefrontal neurons in vitro by increasing potassium conductance, which reduces firing rate20–22 and opposes the effect of AMPA receptor stimulation.90 On the other hand, 5-HT2A receptor activation has been reported to evoke both neuronal excitation and inhibition. 20,91–95 The former action involves an enhancement of AMPA-mediated inputs onto pyramidal neurons,92,93 whereas inhibitory actions are mediated through an increase of synaptic GABA inputs,95 possibly through the activation of 5-HT2A receptors in local-circuit GABAergic interneurons.57
In-vivo actions of 5-HT2A receptor activation in cortical pyramidal neurons
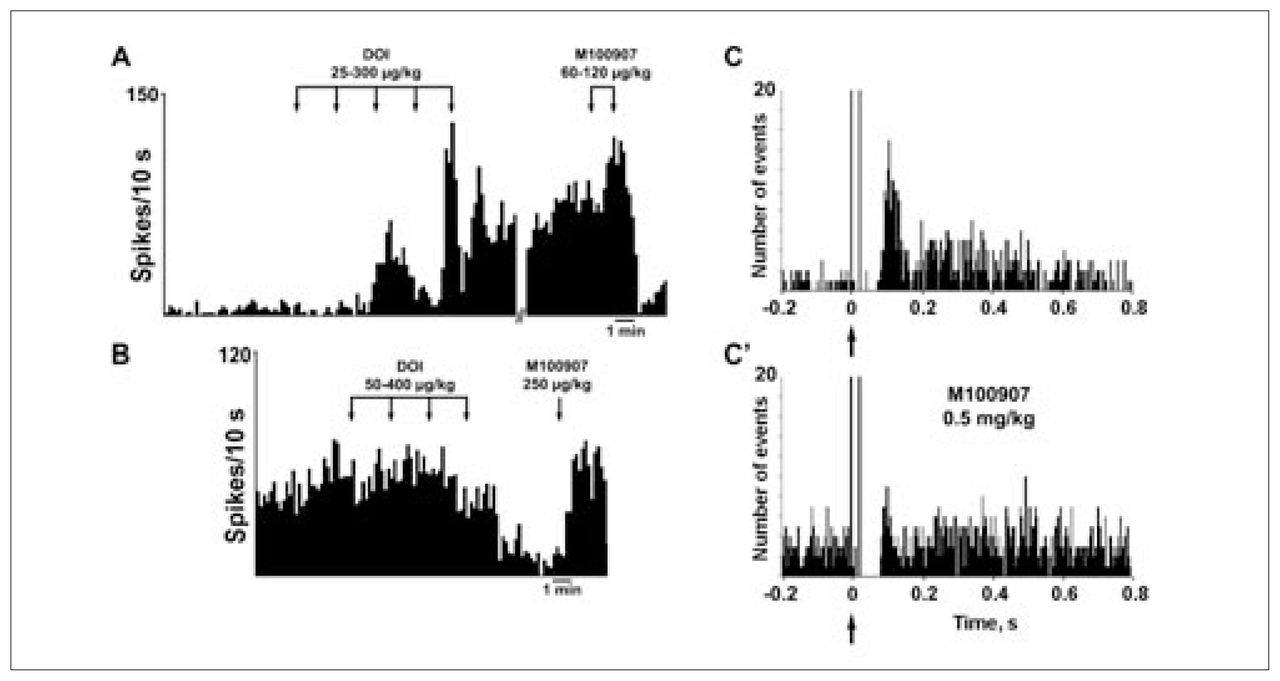
Consistent with the in-vitro studies described earlier, the intravenous administration of the 5-HT2A/2C receptor agonist DOI affected the firing rate of identified pyramidal neurons recorded extracellularly. DOI excited (to 481% of baseline) 38% (21/56) of the neurons recorded, inhibited (to 11% of baseline) 30% (17/56) of the neurons recorded and left the rest unaffected.96 Considering all neurons, DOI increased 2.4-fold the pyramidal firing rate (Fig. 3). These effects were antagonized by the 5-HT2A receptor antagonist M100907.96 Likewise, the electrical stimulation of the DR at a physiologic frequency (0.9 Hz) evoked 5-HT2A-mediated excitations in pyramidal neurons of the mPFC. Peristimulus-time histograms showed the presence of excitations (Fig. 3), which had a mean duration of 80 (standard error of the mean [SEM] 8) ms and had a mean latency of 82 (SEM 8) ms (n = 19).96 On occasion, these excitations were sometimes preceded by short latency inhibitions, as in the neuron shown in Fig. 3. The success rate of orthodromic activation varied between neurons and was 51% (SEM 8%) on average. In most pyramidal neurons examined, orthodromic and antidromic excitations were recorded, showing the existence of a strong reciprocal DR–mPFC interplay (Fig. 3, Fig. 4). As observed for the effect of DOI, the DR-induced excitations were reversed by the intravenous administration of the selective 5-HT2A receptor antagonist M100907 in most instances. The mean success rate dropped from 62% to 15% after the administration of M100907 (n = 10).96
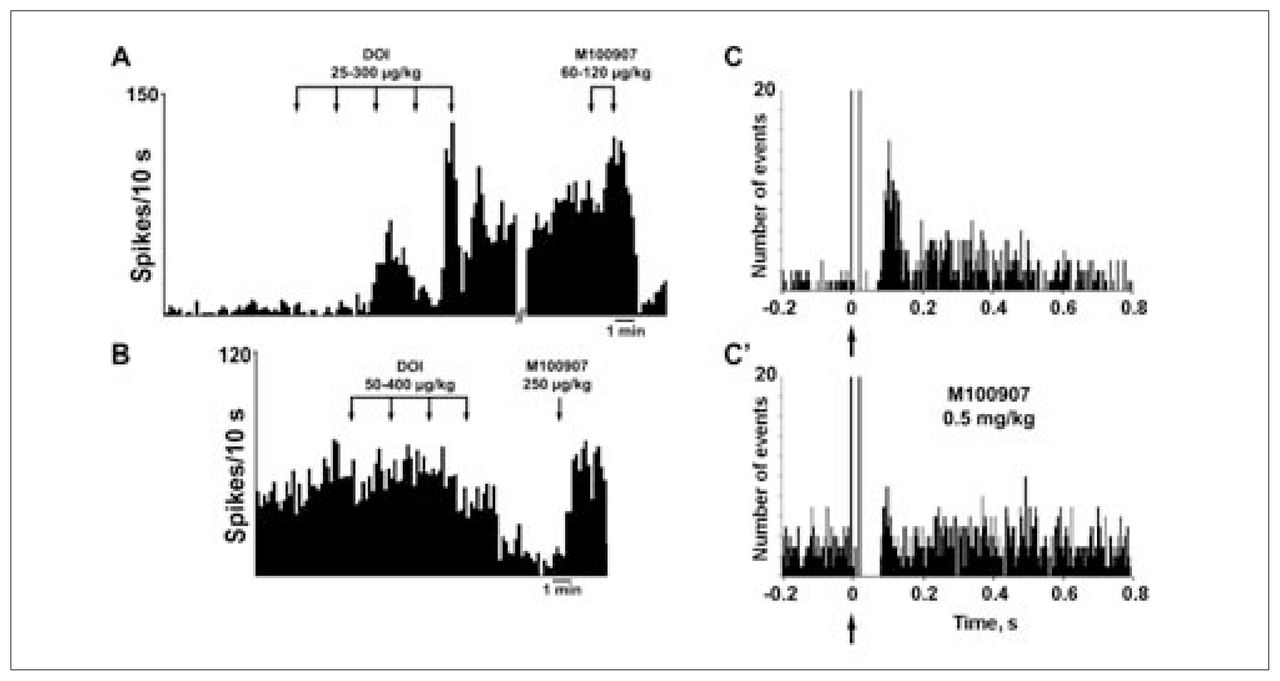
Intravenous administration of the 5-HT2A/2C receptor agonist DOI to anesthetized rats increases (A) and decreases (B) the firing rate of identified pyramidal neurons in the rat mPFC. The proportion of neurons excited, as well as the percent increase, was greater than the proportion of inhibited neurons, which resulted in an overall increase of 240% of the baseline firing rate (n = 56). In most instances, the excitatory and inhibitory effects of DOI were reversed by the selective 5-HT2A antagonist M100907. Panels C and C′ show peristimulus-time histograms of the excitation of a pyramidal neuron in the mPFC evoked by the electrical stimulation of the DR nucleus (0.9 Hz, 0.2-ms square pulses, 1 mA) in basal conditions (C) and after the systemic administration of the selective 5-HT2A antagonist M100907 (500 μg/kg intravenously) (C′). In most instances, the recorded neurons were antidromically activated from the DR or the median raphe nucleus (delay 20 ms), indicating the existence of marked reciprocal interactions between the mPFC and 5-HT neurons in the raphe nuclei. Arrows mark the stimulus artifact. Bin size 4 ms, 170 sweeps. Data from Cereb Cortex 2003;13:870–82.96

Stimulation of the median raphe nucleus inhibits prefrontal pyramidal neurons through the activation of 5-HT1A receptors. In the upper panel, a peristimulus-time histogram shows the presence of an inhibition (latency 32 ms, duration 100 ms) evoked by the electrical stimulation of the median raphe nucleus (0.9 Hz, 0.2-ms square pulses, 2 mA). The lower panel shows the blockade of the inhibition by intravenous administration of the selective 5-HT1A receptor antagonist WAY-100635 (40 μg/kg). Bin size 4 ms, 180 sweeps.
The excitatory effects of DOI appear to involve interaction with glutamatergic transmission. Hence, DOI could increase the excitatory effects of glutamate on prefrontal neurons.91 Likewise, the 5-HT2A receptor-mediated excitatory postsynaptic currents (EPSCs) evoked by 5-HT in layer V pyramidal neurons in rat mPFC in vitro are cancelled by blockade of AMPA receptors and metabotropic glutamate receptor (mGluR) II activation.92,93 Moreover, the modulation of prefrontal NMDA transmission by 5-HT and 1-[2,5-dimethoxy-4-bromophenyl]-2-aminopropane (DOB) appears to involve presynaptic and postsynaptic 5-HT2A receptors.94 Our own data indicate that the selective mGluR II agonist LY-379268 reversed the excitatory effect of DOI on pyramidal neurons in vivo.96 Previous in-vitro studies suggested a role of 5-HT2A receptors putatively located on thalamocortical afferents.93,97 According to this view, 5-HT2A receptor activation would increase glutamate release from thalamic afferents, thus increasing spontaneous EPSCs through the activation of pyramidal AMPA receptors. However, this view is at variance with recent anatomical data indicating that the small proportion of terminal 5-HT2A receptors in the rat mPFC (compared with those in a somatodendritic location) are not located on glutamatergic axons.89 Likewise, our in-vivo data suggest that 5-HT2A receptors responsible for the action of DOI are not located in such terminals, because extensive lesions of the thalamic nuclei projecting to the mPFC do not alter the action of DOI.96 Hence, it is likely that the activation of postsynaptic 5-HT2A receptors in pyramidal neurons mediates the excitatory effect of DOI. Because this action depends on glutamatergic inputs, it may involve a 5-HT2A-mediated synergism with AMPA-mediated transmission (for instance, increasing Ca2+ entry98). These glutamatergic inputs may arise from different cortical or subcortical projections to the mPFC, an issue that deserves further investigation.
In-vivo actions of 5-HT1A receptor activation in cortical pyramidal neurons
Early microiontophoretic studies revealed a predominantly inhibitory action of 5-HT on cortical neurons.7 This effect may involve direct (e.g., 5-HT1A-mediated) or indirect (GABA-mediated) actions of 5-HT.22,91,99 In-vitro intracellular recordings of pyramidal neurons in the PFC suggested that 5-HT1A receptor activation hyperpolarized pyramidal neurons,20,100 possibly by opposing the effects of AMPA-mediated transmission.90 There is limited evidence of the effect of the systemic administration of 5-HT1A agonists on the activity of mPFC neurons. These agents display a biphasic pattern of response, with an initial excitatory phase followed by a reduction of firing rate at higher doses.101 On the other hand, the electrical stimulation of the DR also inhibits presumed pyramidal prefrontal neurons, an effect dependent on the extracellular 5-HT concentration in the mPFC.102 We performed a systematic study of the inhibitory effects of DR and the median raphe nucleus (MnR) stimulation on mPFC pyramidal neurons, identified by antidromic stimulation from these nuclei.103 Fig. 4 shows the inhibitory effect of the stimulation of the MnR on a mPFC pyramidal neuron of a rat anesthetized with chloral hydrate. As previously shown for the DR,102 stimulation of the MnR inhibited the pyramidal neuron, with a short latency and a duration of 100 milliseconds in this particular case. This effect was mediated by 5-HT1A receptors, as shown by the partial reversal exerted by the selective 5-HT1A antagonist WAY-100635.104
5-HT1A–5-HT2A receptor interaction in mPFC pyramidal neurons
Previous reports indicate that 5-HT1A receptor agonists suppress the DOI-induced head shakes, an effect mediated by 5-HT2 receptors.47 Both electrophysiologic20,22 and immunohistochemical59 evidence suggested the co-expression of 5-HT1A and 5-HT2A receptors in the PFC. Therefore, because 5-HT2A receptors excite and those of 5-HT1A inhibit the activity of pyramidal neurons, it is conceivable that the behavioural observations noted earlier are mediated by opposing effects of DOI and 5-HT1A receptor agonists in cortical motor areas. We tested whether such an interaction exists in the mPFC by examining the effects of DOI and 5-HT1A agonists on terminal 5-HT release in the mPFC. As reviewed earlier, the electrical activity of DR 5-HT neurons and the terminal release of 5-HT in the mPFC are under the influence of postsynaptic 5-HT receptors in this area, possibly located on excitatory afferents to the DR.59,67 Although the electrical stimulation can also inhibit 5-HT neurons via GABA interneurons,67,72 it appears that the overall influence of mPFC neurons on serotonergic function is excitatory, because the activation of 5-HT2A and 5-HT1A receptors in the mPFC (with local DOI and 8-OH-DPAT applications, respectively) increased and decreased, respectively, the firing of 5-HT neurons in the DR and the terminal 5-HT release in the mPFC.59,67,96 Hence, the in-vivo 5-HT release in the mPFC may be taken as a surrogate measure of the overall influence of prefrontal inputs to DR 5-HT neurons (Fig. 5), using an experimental model reported elsewhere.59 Figure 5 shows the increase of 5-HT release elicited by the local application of DOI in the mPFC of freely moving rats and the reversal of the effect induced by the co-perfusion of the 5-HT1A agonists BAY x 3702, 8-OH-DPAT, buspirone or ipsapirone. The 5-HT1A-mediated reduction of the DOI-stimulated 5-HT release in the mPFC was antagonized by the earlier treatment of the rats with pertussis toxin, which uncouples the 5-HT1A receptor protein from the potassium channel,105 and by EEDQ, a chelating agent that inactivates several G protein-coupled receptors.106

The local application by reverse dialysis of the 5-HT2A/2C receptor agonist DOI (100 μmol/L) increases the in-vivo 5-HT release in the mPFC through the selective activation of 5-HT2A receptors.59,96 This effect is counteracted by the co-perfusion of various selective 5-HT1A receptor agonists: BAY x 3702, 30 μmol/L; 8-OH-DPAT, 100 μmol/L; or buspirone, 300 μmol/L.104 The opposite action of 5-HT2A and 5-HT1A agonists on 5-HT release in the mPFC is likely to be mediated by changes in the prefrontal inputs onto raphe 5-HT neurons, which subsequently result in parallel changes in terminal 5-HT release.
B: As observed after its systemic administration, DOI is likely to increase the firing rate of pyramidal neurons in the mPFC that project to DR 5-HT neurons (see also Fig. 2). This effect would be counteracted by the 5-HT1A receptor-mediated pyramidal hyperpolarization resulting from the co-application of the 5-HT1A agonists. Atypical antipsychotic drugs and other agents acting as 5-HT2A receptor antagonists may alter the existing balance between 5-HT2A and 5-HT1A and, possibly, other receptors in the mPFC, thus changing the pyramidal output to subcortical structures whose derangements are suspected of underlying depressive symptoms.
Functional consequences and therapeutic implications of 5-HT2A receptor blockade during SSRI treatment
The data outlined here indicate that the generation of nerve impulses in pyramidal neurons of the mPFC is regulated in the opposite manner by postsynaptic 5-HT2A and 5-HT1A receptors. The physiologic and pharmacologic activation of 5-HT2A receptors results in an overall increase in pyramidal activity, whereas that of 5-HT1A receptors inhibits pyramidal activity. The functional interaction between both receptors can be accounted for by their high degree of co-expression (nearly 80%) in the same neuronal populations in the PFC, as assessed by double in situ hybridization,104 which suggests that these interactions occur at the cellular level and are later translated at the circuit level (Fig. 5). Atypical antipsychotic drugs are 5-HT2A receptor antagonists60,61 and behave as functional 5-HT1A receptor agonists.86 Hence, it is possible that they partly exert their therapeutic action by reducing the activity of pyramidal neurons in the mPFC. These project to the ventral tegmental area and control the activity of dopaminergic neurons.63,64,107 Hence, the 5-HT2A-mediated attenuation of the excitatory pyramidal output to subcortical structures may result in a reduction of the activity of ascending dopaminergic neurons. This would reduce the hyperactivity of the mesolimbic pathway108 without concurrently blocking D2 receptors in the nigrostriatal pathway, an action that might explain the lesser extrapyramidal side effects of atypical antipsychotics.
However, the effect of 5-HT2A receptor blockade may be more complex during SSRI treatments, namely, when atypical antipsychotics are used to augment the therapeutic effect of antidepressant drugs. In such conditions, the tone on cortical 5-HT receptors is presumably greater than normal because of the increase in extracellular 5-HT produced by long-term SSRI administration. 30 Second, other 5-HT receptors, such as 5-HT1B, 5-HT2C or 5-HT4/6/7, are present in this brain area, may also control pyramidal cell activity and may therefore additionally modulate the opposite actions of 5-HT1A and 5-HT2A receptors on pyramidal output. The effect of long-term SSRI treatment on these receptors is still poorly known. As mentioned earlier, 5-HT2A receptors in pyramidal and GABAergic neurons are responsible for the excitatory and inhibitory effects of 5-HT, respectively, on pyramidal neurons.92,95 In the basal in-vivo situation, 5-HT2A-mediated excitatory effects appear to predominate,96 yet this balance may be altered by SSRI treatments, which, contrary to the tricyclic drugs, appear to upregulate 5-HT2A receptor binding.109 Clearly, more research is needed to determine how SSRI treatments may alter the balance between 5-HT-mediated excitatory and inhibitory inputs onto prefrontal neurons. This information will improve our understanding of the role played by 5-HT2A receptors in antidepressant treatments, and more specifically, how 5-HT2A receptor blockade may affect the prefrontal circuits involving areas relevant to the treatment of depressive symptoms.
Previous articles in this series
Lesch KP. Gene–environment interaction and the genetics of depression. J Psychiatry Neurosci 2004; 29(3):174–84.
Barden N. Implication of the hypothalamic–pituitary–adrenal axis in the physiopathology of depression. J Psychiatry Neurosci 2004;29(3):185–93.
Malberg J. Implications of adult hippocampal neurogenesis in antidepressant action. J Psychiatry Neurosci 2004;29(3):196–205.
Blier P, Gobbi G, Haddjeri N, Santarelli L, Mathew G, Hen R. Impact of substance P receptor antagonism on the serotonin and norepinephrine systems: relevance to the antidepressant/anxiolytic response. J Psychiatry Neurosci 2004;29(3):208–18.
Acknowledgements
Supported by grant FIS 2001-1147 (Instituto Carlos III; Ministry of Health) and the Fundació La Marató TV3. Some of the reviewed studies, carried out by our group, have been supported by research contracts with Bayer and Eli Lilly. Drs. Amargó-Bosch and Puig are recipients of predoctoral fellowships from the Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS).
Footnotes
Medical subject headings: antidepressive agents; depression; dorsal raphe nucleus; gamma-aminobutyric acid; medial prefrontal cortex; microdialysis; pindolol; receptors, AMPA; serotonin; serotonin uptake inhibitors.
Competing interests: None declared for Drs. Celada, Puig, Amargó-Bosch and Adell. Dr. Artigas has received speaker and consultant fees from Bristol-Myers Squibb.
- Received July 4, 2003.
- Revision received December 11, 2003.
- Accepted December 16, 2003.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.