

Abstract
Background: The hemizygous 22q11.2 microdeletion is a common copy number variant in humans. The deletion confers high risk for neurodevelopmental disorders, including autism and schizophrenia. Up to 41% of deletion carriers experience psychotic symptoms.
Methods: We present a new mouse model (Df(h22q11)/+) of the deletion syndrome (22q11.2DS) and report on, to our knowledge, the most comprehensive study undertaken to date in 22q11.2DS models. The study was conducted in male mice.
Results: We found elevated postpubertal N-methyl-d-aspartate (NMDA) receptor antagonist–induced hyperlocomotion, age-independent prepulse inhibition (PPI) deficits and increased acoustic startle response (ASR). The PPI deficit and increased ASR were resistant to antipsychotic treatment. The PPI deficit was not a consequence of impaired hearing measured by auditory brain stem responses. The Df(h22q11)/+ mice also displayed increased amplitude of loudness-dependent auditory evoked potentials. Prefrontal cortex and dorsal striatal elevations of the dopamine metabolite DOPAC and increased dorsal striatal expression of the AMPA receptor subunit GluR1 was found. The Df(h22q11)/+ mice did not deviate from wild-type mice in a wide range of other behavioural and biochemical assays.
Limitations: The 22q11.2 microdeletion has incomplete penetrance in humans, and the severity of disease depends on the complete genetic makeup in concert with environmental factors. In order to obtain more marked phenotypes reflecting the severe conditions related to 22q11.2DS it is suggested to expose the Df(h22q11)/+ mice to environmental stressors that may unmask latent psychopathology.
Conclusion: The Df(h22q11)/+ model will be a valuable tool for increasing our understanding of the etiology of schizophrenia and other psychiatric disorders associated with the 22q11DS.
Introduction
The 22q11.2 hemizygous microdeletion confers very high risk for neurodevelopmental disorders, including autism and schizophrenia (22q11.2 deletion syndrome [22q11.2DS]). The estimated prevalence is approximately 1 in 2000.1 The International Consortium on Brain and Behaviour in 22q11.2 has recently reported the cumulated prevalence of schizophrenia to be 24% in adolescence and 41% in adulthood.2 Studies of patients with schizophrenia find that 22q11.2 deletion accounts for approximately 0.3% of the cases.3
Despite massive efforts there is still no coherent understanding of the etiology of schizophrenia — a highly heritable heterogeneous disorder with strong environmental influence.4,5 Several neurotransmitters are implicated in the disorder: glutamate,6 γ-aminobutyric acid (GABA),7 dopamine (DA)8 and acetylcholine signalling9 have all been highlighted in the disease etiology and manifestation. The cognitive impairment and negative symptomatology have been related to dysfunction in regulation of glutamate–GABA transmission leading to excitatory–inhibitory imbalances.7 Like in individuals with schizophrenia,10,11 cognition12 and information processing is disrupted in children with 22q11.2 deletion, in whom schizophrenia has not (yet) developed.13,14 Given the highly increased risk for schizophrenia and the phenotypic overlap between schizophrenia and the 22q11.2DS, studying the consequence of the 22q11.2 deletion provides a unique opportunity to add to the understanding of the etiology of schizophrenia and other related psychiatric disorders, which eventually may lead to novel drugs targeting the core of the disease.
Five transgenic mouse models of the 22q11.2DS have been generated by different research groups (Table 1). These models have recently been reviewed by Hiroi and colleagues,20 who also provide an overview of key genes in the 22q11 region. Inconsistent results have been reported from assays addressing cognitive functions relevant to schizophrenia (unpublished observations), whereas studies of other symptom domains have been more consistent (Table 1). Decreased prepulse inhibition (PPI) and increased acoustic startle response (ASR) have been consistently observed (LgDel,16 Df1/+17 and Df(16)A+/−18). However, hearing was recently shown to be impaired in the Df1/+ mouse,19 raising the possibility that hearing loss rather than altered sensorimotor gating might underlie PPI deficits in 22q11DS mouse models.
Summary of results for Df(h22q11)/+ and other 22q11.2DS mouse models
The LgDel,16 Df1/+17 and Df(16)A+/−18 mice used in the referred studies were noncongenic. Genetic background21 and environmental factors22 may influence phenotypic expression. To be able to control for these factors and ensure sufficient access to mice we have generated a new congenic mouse model (Df(h22q11)/+) of the 22q11.2DS as part of a large multisite collaboration. Here we report on an extensive characterization of these mice using a set of assays related to the pathophysiology of schizophrenia. The Df(h22q11)/+ mice display phenotypes relevant for modelling aspects of schizophrenia-related symptoms, including increased postpubertal N-methyl-d-aspartate (NMDA) antagonist sensitivity and age-independent PPI and ASR deficits. These impairments were not a result of impaired hearing. The Df(h22q11)/+ mice also display increased amplitude of loudness-dependent auditory evoked potentials (LDAEP), prefrontal cortex (PFC) and dorsal striatal elevations of the DA metabolite dihydroxyphenylacetic acid (DOPAC), and increased dorsal striatal expression of the AMPA receptor subunit GluR1. The Df(h22q11)/+ mice did not deviate from wild-type (WT) mice in a range of other behavioural and biochemical assays.
Methods
Animals
The Df(h22q11)/+ mouse line was generated by TaconicArtemis. Animals were bred by mating WT C57BL/6N females with hemizygotic Df(h22q11)/+ males to avoid any placental or maternal care effects of the deletion. Animals were weaned at 3 weeks, and tail biopsies were collected for polymerase chain reaction (PCR)–based genotyping. Mice were group-housed (2 WT mice and 2 hemizygotes from the same litter per cage) under controlled laboratory conditions (12-h light–dark cycle, 20 ± 2°C, 30%–70% humidity) in standard mouse cages with sawdust bedding, environmental enrichment (plastic house and paper for nesting), and food and water available ad libitum. After surgery mice were single-housed (see Appendix 1, available at jpn.ca). All experiments were carried out using littermate controls. The experiments used 27 cohorts (n = 12–95) of male mice aged 6–26 weeks (Appendix 1, Fig A2). The selection of cohorts and animals for individual experiments was not randomized. All studies were carried out in accordance with the local legislation based on the European Union regulation (directive 2010/63 of Sept. 22, 2010) and UK Animals (Scientific Procedures) Act of 1986. The studies were approved by the Barcelona School of Medicine Institutional Animal Care and Use Committee or the Danish National Committee for Ethics in Animal Experimentation.
Drugs
Clozapine (obtained from Novartis) was dissolved in 0.1 M of hydrochloride and diluted with saline. Gabazine (SR95531, obtained from Sigma-Aldrich) was dissolved in 0.2 M of NaCl. Haloperidol (obtained from Sigma) was dissolved in 0.1 M of tartaric acid and diluted with saline. Phencyclidine (PCP) hydrochloride (synthesized by Lundbeck) was dissolved in 0.1 M of methanesulfonic acid and diluted with saline. (S)-(+)-ketamine (obtained from Sigma Aldrich) was dissolved in 5% (w/v) glucose solution at a concentration of 10 mg/mL.
For the microdialysis studies, veratridine and PCP were purchased from Sigma-Aldrich and nomifensine from Tocris. Veratridine was dissolved in DMSO (5 mM) and nomifensine (1 mM) in artificial cerebrospinal fluid (aCSF).
Basal characterization
Animals were characterized in the hot plate, rotarod, beam-walk, bright open field, locomotor activity and elevated plus maze.
Prepulse inhibition of the ASR
The procedure has been extensively described elsewhere.23 Six cohorts of animals were tested without drug treatment (age 6–21 wk). Two cohorts were treated with either clozapine (10 mL/kg administered subcutaneously at age 13 wk) or haloperidol (10 mL/kg administered subcutaneously at age 15 wk).
NMDA receptor antagonist–induced hyperactivity
Following a 60-min habituation phase to a novel environment, Df(h22q11)/+ and WT littermates were treated with PCP or S(+)-ketamine (10 mL/kg administered subcutaneously), and their activity was monitored for a further 60 min.
Anatomy
We conducted gross brain characterization, including assessment of brain weight, ventricular size, hippocampal structure, myelination, cortical thickness and layer composition, using cortical NeuN and parvalbumin immunoreactivity and solochrome staining.
Western blotting
We carried out a targeted screen (Peggy–Simple western blotting) for proteins in the PFC, sorsal striatum and hippocampus. Markers showing significant changes across genotypes were replicated twice. The first replication used new dilutions from each of the 11 samples per genotype separately, whereas the second replication used 1 pooled sample for each genotype, which was run twice.
Tissue content measured using high-performance liquid chromatography
We collected and analyzed PFC and dorsal striatal tissue content of DA, DOPAC, homovanillic acid (HVA), noradrenaline (NA), 5-hydroxytryptamine (5-HT) and 5-hydroxyindoleacetic acid (5-HIAA) as previously described.24
Microdialysis in freely moving animals
We assessed the functional state of dorsal striatal DA transmission as previously described.25 Briefly, after collection of baseline DA dialysate fractions, extracellular DA levels were measured following reverse dialysis by application of the depolarizing agent veratridine (50 mM, local 20-min pulse) and the DA/NA transporter uptake inhibitor nomifensine (50 mM, local application during 8 fractions). We subcutaneously administered PCP (2.5 + 2.5 mg/kg) systemically in the same animals the following day. Microdialysis measures were taken +0.5 mm anteroposterior (AP), −1.7 mm medial lateral (ML) and −4.5 mm dorsal ventral (DV) from Bregma.26
Electrophysiology
Loudness dependent auditory evoked potentials
Auditory stimuli (white noise, 5 ms duration with 1 ms rise and fall) were presented to awake freely moving mice at different intensity levels (60, 70, 80, 90 and 100 dB) with a 6-s interstimulus interval. We obtained 20-min electroencephalography (EEG) recordings during auditory stimulation. Grand average AEPs from the auditory cortex (AuC) were constructed consisting of a 100-ms prestimulus baseline and a 900-ms poststimulus interval. The peak N1 and P2 amplitudes were determined as the most negative deflection 10–25 ms poststimulus and the most positive deflection 22–65 ms poststimulus, respectively. Peak-to-peak N1/P2 amplitudes were determined at each intensity. We calculated linear regression between peak-to-peak N1/P2 amplitude and sound intensity, and the LDAEP was determined as the mean slope of the linear regression.
Auditory-induced brainstem response
Auditory-induced brainstem responses (ABRs) were recorded with auditory stimulation to the left and right ears of anaesthetized animals with hollow ear bars guiding sound waves to the external auditory meatus. Three subdermal electrodes were inserted; the active electrode was placed at the vertex, the reference electrode was placed at the ear being tested, and the ground electrode was placed near the opposite ear. We recorded the ABRs during a series of click stimuli (duration: 50 μs, rate: 20 Hz/s, intensity: 30–100 dB in 5-dB steps) with 500 repeats per intensity level. Stimuli were presented to 1 ear, with the nonstimulated ear being blocked with an earplug. At each intensity ABRs were averaged and analyzed within a 7-ms poststimulus window. We identified ABR thresholds by visual inspection blinded to the genotype for both left and right ears and defined the thresholds as the lowest intensity evoking a deflection of the ABR wave. The ABR protocol used was modified from that used by Fuchs and colleagues.19
Low-frequency cortical oscillation
Medial prefrontal cortical (mPFC) low-frequency oscillation (LFO) power was assessed at baseline and following systemic PCP treatment (10 mg/kg administered subcutaneously) as previously described.27 Recordings were made at +2.1 mm AP, −0.2 to −0.4 mm ML and −1 to −2.5 mm DV from Bregma.26
Medial PFC GABAA receptor function
We assessed mPFC GABAA receptor (GABAAR) function by examining discharge rates of putative pyramidal neurons in control conditions and during local application of the GABAAR antagonist gabazine as previously described.28 Recordings were made at +2.1 mm AP, −0.2 to −0.4 mm ML and −1 to −2.5 mm DV from Bregma.26
Statistical analysis
We performed our statistical analyses using SigmaPlot version 11.2 (Systat Software Inc.) or SPSS version 21.0 (IBM Corp). Data were analyzed using either 2-way analysis of variance (ANOVA), 2-way repeated-measures ANOVA, or Student t test. We conducted post hoc tests using the Holm–Sidak method, and for the t test we used the Mann–Whitney rank sum test if the normality (Shapiro–Wilk) or equal variance (Levene) test failed.
No adjustments were made for multiple testing beyond the post hoc tests. Data were visually inspected for outliers. No obvious outliers were identified, and all data were included in the analysis. We had only a few missing values, which we handled in SigmaPlot using a general linear model.
Results
Generation of mice and basic phenotyping
The Df(h22q11)/+ mouse line was generated by deletion of the human 22q11.2DS orthologous genomic region on mouse chromosome 16 (Fig. 1A and Appendix 1, Fig. A1). Expression levels of deleted genes in cortical areas were examined using microarray analysis (Fig. 1B). Except for Tssk1, Vpreb2, and Cdc45, expression of all detected genes in the deleted region were significantly reduced to roughly 50% of the expression in WT mice. Absolute expression levels assessed by RNAseq analysis of cortex in WT mice showed that Tssk1, Vpreb2, and Cdc45 along with the undetected Tssk2 and Gsc2 genes were very lowly expressed, suggesting that the apparent unchanged expression of these genes are due to very low signal-to-noise ratio in the micro-array detection (data not shown). Expression of the flanking gene Car15 was also reduced by 50% in Df(h22q11)/+ mice, whereas expressions of other flanking genes were unaltered. Only around 40% of newborn pups were hemizygous, indicating reduced conception or intrauterine survival (Df(h22q11)/+ fraction ~0.40, p = 0.042; Appendix 1, Fig. A3). In the studies that followed, hemizygous males were compared with WT male littermates.
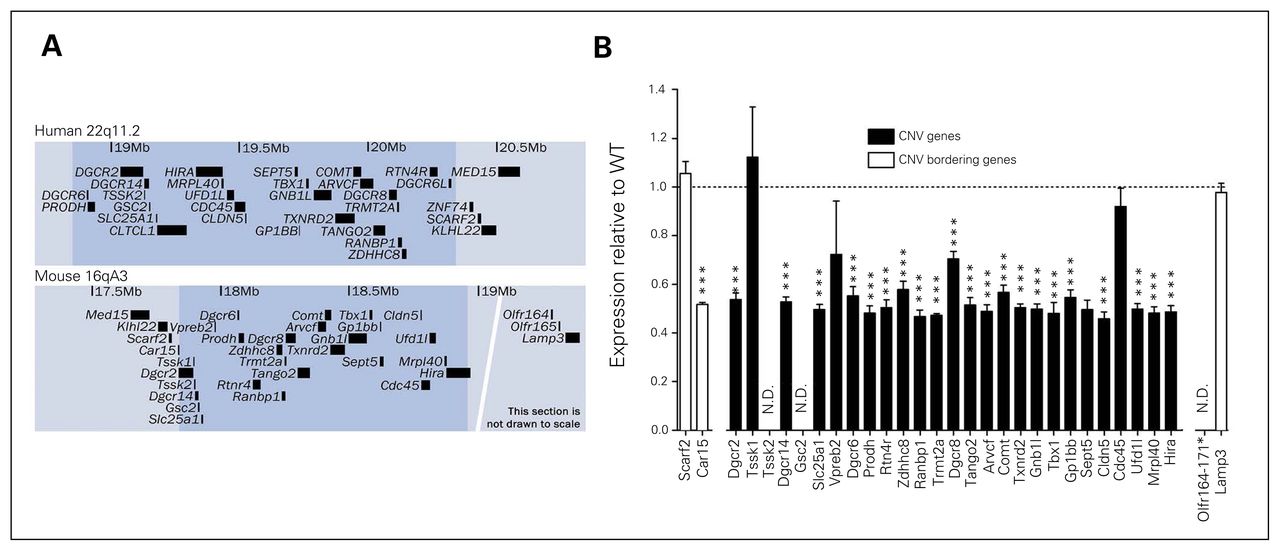
Construct similarities in Df(h22q11)/+ and human deletion carriers. (A) Overview of the deleted region (shaded) in humans (22q11.2) and the corresponding orthologous region in mice (16qA3). The maps are based on human library GRCh38/hg38 and mouse library GRCm38/mm10 from the University of California, Santa Cruz (UCSC) database. Only annotated RefSeq sequences are shown for clarity. (B) Median normalized microarray analysis of relative cortical expression of gene products from deleted segment in Df(h22q11)/+ mice compared with wild-type (WT) littermates. *Significant difference between WT and Df(h22q11)/+ mice (***p < 0.001). CNV = copy number variation; N.D. = not detected.
Overall, Df(22q11.2)/+ mice appeared healthy with grossly normal behaviour, including pain sensitivity (hotplate: 50–56°C), motor function (beam-walking, locomotor activity, rotarod) and anxiety response (bright open field, elevated plus maze; Appendix 1, Fig. A3 ), and with normal brain weight and gross morphology (cortical layer composition, hippocampal structures, parvalbumin positive interneuron counts and myelin patterns; Appendix 1, Fig. A3 and A4).
Behaviour
Prepulse inhibition
We found that PPI was significantly reduced in Df(h22q11)/+ mice (Fig. 2A). Data were collapsed across prepulse intensities, as there was no genotype × prepulse intensity (5, 10, 15 dB) interaction. The PPI impairment was stable during development and observed consistently in 6 independent cohorts of 6- to 21-week-old animals. The impairment was resistant to treatment with haloperidol (Fig. 2B) and clozapine (Fig. 2C), representing typical and atypical antipsychotics, respectively. Clozapine impaired PPI in the WT mice at the highest dose tested (2 mg/kg).

Behavioural characterization of Df(h22q11)/+ mice and wild-type (WT) littermates. (A) Df(h22q11)/+ mice showed decreased prepulse inhibition (PPI). The PPI deficit was reproduced in 6 independent cohorts of mice 6–21 weeks old (n = 12–24/group). We conducted a t test for each week (week 6: t45 = 3.6, p < 0.001; week 9: t30 = 4.8, p < 0.001; week 15: t29 = 6.3, p < 0.001; week 17: t22 = 3.9, p < 0.001; week 19: t22 = 4.9, p < 0.001; week 21: t22 = 6.4, p < 0.001). (B) Haloperidol did not rescue the PPI deficit (n = 12/group, 2-way analysis of variance [ANOVA], genotype: F1,94 = 69.1, p < 0.001; dose: F3,94 = 1.02, p = 0.38; genotype × dose: F3,94 = 0.59, p = 0.63). (C) Clozapine did not rescue the PPI deficit (n = 13/group, 2-way ANOVA, genotype: F1,87 = 46.3, p < 0.001; dose: F3,87 = 4.87, p = 0.004; genotype × dose: F3,87 = 6.14, p < 0.001). (D) Df(h22q11)/+ mice had increased acoustic startle response (ASR). The increased ASR was found in 6 independent cohorts of mice 6–21 weeks old (n = 12–24/group). We conducted a t test for each week (week 6: Tn23,24 = 418, p = 0.004; week 9: Tn16,16 = 204, p = 0.025; week 15: Tn15,16 = 340, p < 0.001; week 17: Tn12,12 = 79, p < 0.001; week 19: t22 = −4.51, p < 0.001; week 21: Tn12,12 = 101, p = 0.005)). (E) Haloperidol did not attenuate the increased ASR (n = 12/group, 2-way ANOVA, genotype: F1,94 = 88.5, p < 0.001; dose: F3,94 = 0.060, p = 0.98; genotype × dose: F3,94 = 0.13, p = 0.95). (F) Clozapine did not attenuate the increased ASR (n = 13/group, 2-way ANOVA, genotype: F1,87 = 83.1, p < 0.001; dose: F3,87 = 9.35, p < 0.001; genotype × dose: F3,87 = 1.49, p = 0.22). (G) Age-dependent hypersensitivity to phencyclidine (PCP)-induced locomotion in Df(h22q11)/+ mice (n = 12/group). We conducted a t test for each week (week 7: t22 = 1.10, p = 0.28 at 2.5 mg/kg and t22 = −0.68, p = 0.50 at 5.0 mg/kg; week 9: Tn11,12 = 90, p = 0.011 for vehicle; Tn12,12 = 187, p = 0.035 at 1.25 mg/kg; t22 = −2.33, p = 0.029 at 2.5 mg/kg and t22 = −0.91, p = 0.37 at 5.0 mg/kg). (H) Hypersensitivity to ketamine-induced locomotion in Df(h22q11)/+ mice (n = 13–16/group, 2-way ANOVA, genotype: F1,54 = 0.23, p = 0.64; dose: F11,54 = 25.6, p < 0.001; genotype × dose: F1,54 = 5.95, p = 0.018; post hoc test within ketamine 10 mg/kg: t = 2.05, p = 0.044). Data are presented as mean ± standard errors of the mean (SEM). *Significant differences between WT and Df(h22q11)/+ mice (*p < 0.05, **p < 0.01, ***p < 0.001). #Significant difference within genotype relative to vehicle (#p < 0.05, ###p < 0.001).
Acoustic startle response
Df(h22q11)/+ mice had increased ASR. This was observed in 6 independent cohorts of 6- to 21-week-old animals (Fig. 2D). Haloperidol did not attenuate the increased ASR (Fig. 2E). Clozapine decreased ASR in both WT and Df(h22q11)/+ mice. However, no genotype × treatment interaction effect was observed (Fig. 2F).
PCP-induced hyperactivity
Df(h22q11)/+ mice showed increased activity to PCP-induced locomotor activity in an age-dependent manner. The hyperreactivity was seen in 9-week-old but not 7-week-old mice (Fig. 2G).
S-(+)-ketamine-induced hyperactivity
Df(h22q11)/+ mice at 26 weeks of age showed hyperreactivity to ketamine-induced locomotor activity (Fig. 2H).
Biochemistry
Tissue content
Df(h22q11)/+ mice had increased levels of DOPAC in the PFC (Fig. 3A) and dorsal striatum (Fig. 3B), whereas the levels of DA, HVA, NA, 5-HT and 5-HIAA were unaffected.
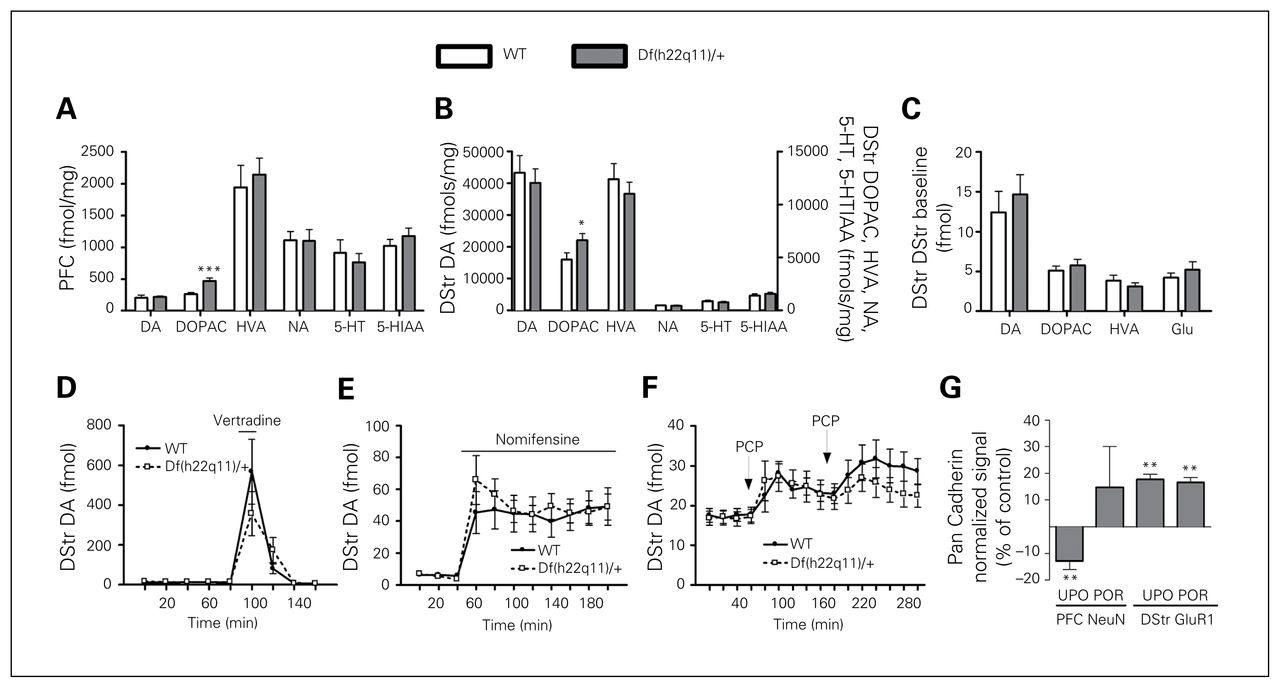
Biochemical measures in Df(h22q11)/+ mice and wild-type (WT) littermates. (A) Whole tissue content in the prefrontal cortex (PFC). The level of 3,4-dihydroxyphenylacetic acid (DOPAC) was increased in Df(h22q11)/+ mice (n = 8–10/group). We conducted a t test for each analyte (dopamine [DA]: t16 = −0.22, p = 0.83; DOPAC: t16 = −3.85, p = 0.001; homovanillic acid [HVA]: t16 = −0.48, p = 0.64; noradrenaline [NA]: Tn8,10 = 81, p = 0.69; 5-hydroxytryptamine [5-HT]: Tn8,10 = 83, p = 0.56; 5-hydroxyindoleacetic acid [5-HIAA]: t16 = −0.93, p = 0.38). (B) Dorsal striatum (DStr) whole tissue content. The level of DOPAC was increased in Df(h22q11)/+ mice (n = 9–15/group). We conducted a t test for each analyte (DA: t27 = 0.45, p = 0.66; DOPAC: Tn14,15 = 156, p = 0.020; HVA: Tn14,15 = 215, p = 0.84; NA: Tn15,15 = 238, p = 0.85; 5-HT: t28 = 0.91, p = 0.37; 5-HIAA: Tn15,15 = 198, p = 0.16). (C) No genotype effect was detected in the microdialysis of the DStr at baseline (n = 14–15/group). We conducted a t test for each analyte (DA: T13,15 = 203, p = 0.52; DOPAC: t27 = −0.69, p = 0.50; HVA: t19 = 0.98, p = 0.34; glutamate [Glu]: T14,14 = 191, p = 0.58). (D) Veratridine (VTD) increased DStr DA similarly in WT and Df(h22q11)/+ mice (n = 14/group, 2-way repeated-meadures ANOVA, genotype: F1,26 = 0.24, p = 0.63; time: F8,208 = 18.8, p < 0.001; genotype × time: F8,208 = 1.35, p = 0.22). (E) Nomifensine increased DStr DA similarly in WT and Df(h22q11)/+ mice (n = 14/group, 2-way repeated-measures ANOVA, genotype: F1,26 = 0.12, p = 0.74; time: F10,260 = 25.5, p < 0.001; genotype × time: F10,260 = 0.79, p = 0.64). (F) PCP increased DStr DA similarly in WT and Df(h22q11)/+ mice (n = 14/group) following both the first (genotype: F1,28 = 0.003, p = 0.958; genotype × time: F4,112 = 0.742, p = 0.565) and second PCP challenge (genotype: F1,28 = 1.632, p = 0.212; genotype × time: F5,140 = 0.432, p = 0.826). (G) The initial Western blotting showed decreased PFC NeuN (t20 = 2.92, p = 0.009) and increased DStr GluR1 expression (t20 = 2.99, p = 0.007) in Df(h22q11)/+ mice (n = 11/group). However, the NeuN decrease failed to replicate when samples were pooled. Data are presented as means + standard errors of the mean (SEM). *Significant differences between WT and Df(h22q11)/+ mice (*p < 0.05, **p < 0.01). POR = pooled replication; UPO = unpooled.
Microdialysis in freely-moving animals
Df(h22q11)/+ and WT mice did not differ in baseline dorsal striatal DA, DOPAC, HVA or glutamate levels (Fig. 3C). Local administration of the sodium channel opener veratridine (Fig. 3D) and the DA/NE reuptake inhibitor nomifensine (Fig. 3E) increased DA equally in Df(h22q11)/+ and WT mice. Similarly, PCP increased dorsal striatal DA equally in Df(h22q11)/+ and WT mice following both the first and second PCP challenge (Fig. 3F).
Western blotting
Df(h22q11)/+ mice showed decreased PFC NeuN and increased dorsal striatal GluR1 levels (Fig. 3G). In a replication study using pooled samples from WT and Df(h22q11)/+ mice, respectively, the dorsal striatal GluR1 increase was confirmed while the PFC NeuN signal failed to replicate. There were no effects of genotype on levels of the 14 additional markers measured (Table 2).
Protein level in Df(h22q11)/+ mice and wild-type littermates
Electrophysiology
Auditory induced brainstem response threshold
Df(h22q11)/+ and WT mice did not differ in ABR threshold (Fig. 4A). The ABR threshold did not exceed the previously defined hearing deficit criterion (55 dB SPL).19
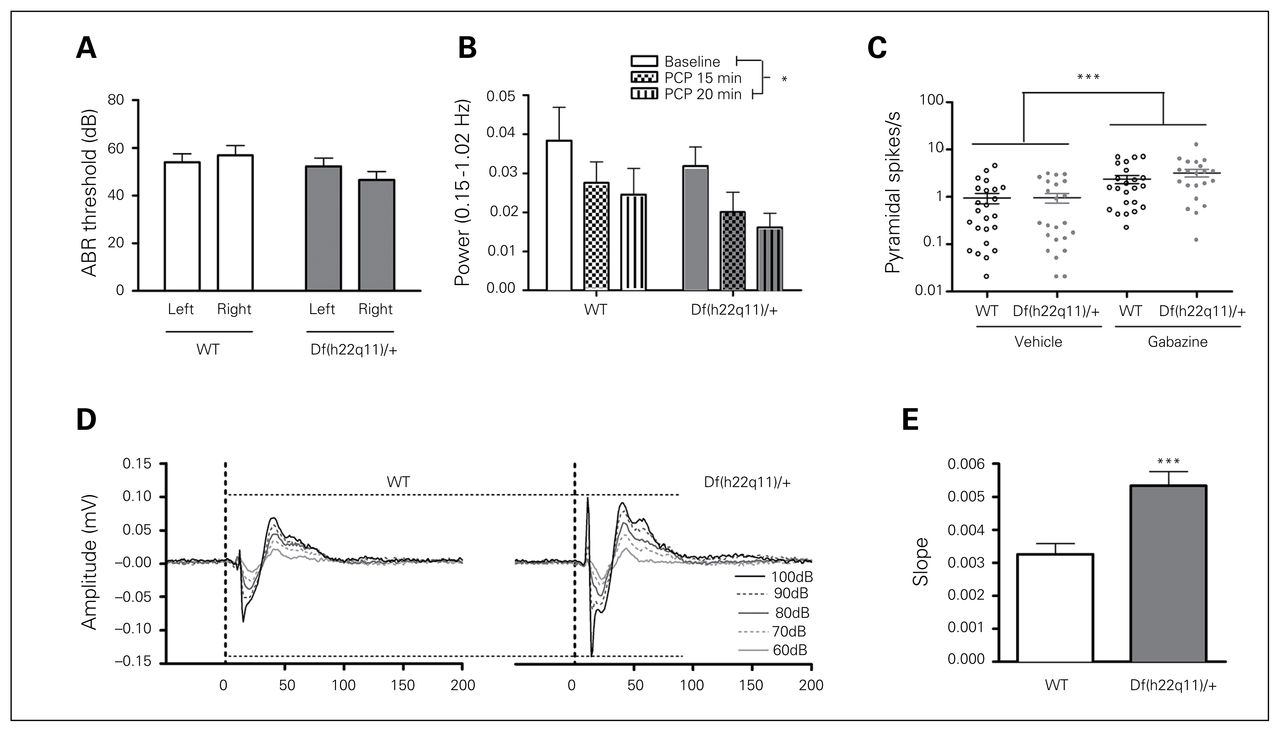
Electrophysiological measures in Df(h22q11)/+ and wild-type (WT) littermates. (A) Hearing did not differ between the Df(h22q11)/+ mice and WT littermates (n = 13/group, 2-way repeated-measures ANOVA, genotype: F1,24 = 1.649, p = 0.211; ear: F1,24 = 0.331, p = 0.571; ear × genotype: F1,24 = 3.570, p = 0.071). (B) Phencyclidine (PCP) caused similar decrease of cortical low-frequency oscillations (LFOs) in WT and Df(h22q11)/+ mice (n = 9/group, 2-way ANOVA, genotype: F1,48 = 2.40, p = 0.13; condition: F2,48 = 3.44, p = 0.040; genotype × condition: F2,24 = 0.013, p = 0.99). (C) Gabazine caused a similar increase in pyramidal spike frequencies in WT and Df(h22q11)/+ mice (n = 14/group, 2-way ANOVA; genotype: F1,87 = 1.21, p = 0.28; treatment: F1,87 = 20.9, p < 0.001; genotype × treatment: F1,87 = 1.12, p = 0.29). (D) Grand average loudness-dependent auditory evoked potentials (LDAEP) waveforms from the auditory cortex (AuC) in WT and Df(h22q11)/+ mice (n = 22–23/ group, 2-way repeated-measure ANOVA, P1/N1 genotype: F1,43 = 22.6, p < 0.001; intensity: F4,43 = 75.5, p < 0.001; genotype × intensity: F4,43 = 13.7, p < 0.001; N1/P2 genotype: F1,43 = 9.89, p = 0.003; intensity: F4,43 = 156.4, p < 0.001; genotype × intensity: F4,43 = 9.76, p < 0.001). (E) Collapsed LDAEP slopes for Df(h22q11)/+ and WT mice (n = 22–23/group, t test; t43 = −3.896, p < 0.001). Data are presented as means ± standard errors of the mean. *Significant differences (*p < 0.05, ***p < 0.001). ABR = auditory-induced brainstem response.
Loudness dependent auditory evoked potentials
The Df(h22q11)/+ mice showed increased LDAEPs in AuC (Fig. 4D and E). There was a significant increase in P1/N1 and N1/P2 amplitudes. The Df(h22q11)/+ mice showed increased LDAEP at higher sound intensities, and the N1/P2 amplitudes were increased at 90 dB (p < 0.001) and 100 dB (p < 0.001). Similar results were generated for other cortical brain regions, with differences in N1 amplitude as the main contributing component (data not shown). There was no effect of genotype on latency at any sound intensity (data not shown).
Medial PFC LFOs
There was no difference between Df(h22q11)/+ and WT mice on baseline or PCP-induced attenuation of cortical LFOs (Fig. 4B). Administration of PCP decreased cortical LFOs.
Medial PFC GABAAR function
There was no difference between Df(h22q11)/+ and WT mice on baseline or GABAAR antagonist–induced elevation of pyramidal neuron spike frequency (Fig. 4C).
Discussion
In this paper we report on the generation and characterization of a new 22q11DS mouse model with a focus on assays relevant for the pathophysiology of schizophrenia. The Df(h22q11)/+ mice had age-independent and antipsychotic-resistant gating deficits measured by PPI. They had increased ASR and increased amplitude of LDAEP. Also, the Df(h22q11)/+ mice exhibited increased NMDAr antagonist–induced locomotion, but in contrast to PPI, this effect was observed only after puberty. The 22q11.2 deletion resulted in increased dorsal striatal and PFC DOPAC levels, as would be expected by the deletion of the Comt gene. Among other markers explored (Fig. 3 and Table 2), we saw a reduction of dorsal striatal GluR1 expression.
Prepulse inhibition deficits and ASR changes have previously been reported in other 22q11DS mouse models.16–18 We confirmed these findings in our model and further examined the age dependency, response to antipsychotic treatment and possible hearing confounds. We found that the PPI and ASR changes in Df(h22q11)/+ mice are robust (observed in 6 independent cohorts), are present before puberty and persist across ages. This effect mirrors the reduced PPI found in human 22q11.2 deletion carriers, which precedes onset of puberty and the potential development of schizophrenia symptoms.13,14 Chronic middle ear infection and elevated click-response ABR threshold have been observed in the Df1/+ mouse model19 raising the possibility that hearing loss at low intensities rather than sensorimotor gating deficits per se underlie the PPI deficits found in the 22q11DS mouse models. We addressed this by measuring ABR thresholds and found those to be unchanged in the Df(h22q11)/+ mice. This, together with the increased amplitude of AEPs found in the LDAEP assay, indicates that hearing is not reduced in the Df(h22q11)/+ mouse. Thus, the decrease in sensorimotor gating observed in the Df(h22q11)/+ mouse is not caused by hearing loss. The contrasting ABR findings between the Df(h22q11)/+ and Df1/+ mouse models19 may be due to differences in strain or environmental conditions affecting the risk of ear infection.
The PPI and ASR impairments were resistant to anti-psychotic treatment (haloperidol and clozapine), which is in overall agreement with the effect of these drugs in the clinical setting. In patients with schizophrenia, classical antipsychotics working through D2/D3 receptor blockade reduce the psychotic symptoms without affecting PPI deficits.29,30 The findings for the new generation of antipsychotics with multi-receptor profiles are more mixed,29,31,32 and a cross-sectional study by Kumari and colleagues33 provided indirect evidence of clozapine being superior to haloperidol for PPI deficits in patients with schizophrenia, which is not reflected by the present data from the Df(h22q11)/+ mice.
Similar to the Df1/+ mouse,34 we found that Df(h22q11)/+ mice displayed increased locomotor activity in response to the NMDAr antagonists PCP and ketamine. Interestingly, the effect was age-dependent and only was observed after puberty (9- but not 7-week-old animals). As schizophrenia is characterized by onset of overt symptoms during or after puberty35 and glutamatergic dysfunctions,36,37 our data suggest that the Df(h22q11)/+ mouse may model aberrant NMDAr-related neurodevelopment trajectories associated with the disorder. The prepubertal and age-independent PPI deficits found in the Df(h22q11)/+ mice suggest that these sensorimotor gating deficits are due to biological perturbation other than what drives the NMDAr antagonist phenotype.
Biochemical assays revealed that the Df(h22q11)/+ mice had elevated PFC and dorsal striatal tissue content of the DA metabolite DOPAC. This phenotype may be explained by the haploinsufficiency for COMT, which catalyzes the demethylation of DA. The role of COMT is well established in low–DAT density brain regions, such as the PFC,38,39 whereas there are contrasting findings in the striatum.40,41 The present data also support a role of COMT in striatal DA function. Despite the reduced expression of COMT and consequent increase in DOPAC we saw no changes in tissue content of DA and HVA, NA or 5-HT. The Df(h22q11)/+ mice also showed normal dorsal striatal DA release following PCP administration and local application of veratridine or nomifensine. Dopamine release is involved in, but temporally dissociated from, NMDA antagonist–induced locomotor activity.42 The observed hypersensitivity to NMDAr blockade is likely not caused by hypersensitivity of the DA system in the Df(h22q11)/+ mice, as release of DA is unchanged. This is further supported by the lack of locomotor hyperresponsivity to amphetamine in these mice (unpublished data). GluR1 was the only glutamatergic-related marker (GluR2, NR2A, NR2B, or VGluT1) explored that showed differential expression, and we did not observe any changes in GABA-related markers (GABAA α1, KCC2, VGAT, or GAD 65/67). Furthermore, PFC GABAergic–glutamatergic interaction measured as GABAAR antagonism–induced pyramidal cell discharge rates was not affected, and measures of PCP-induced attenuation of LFOs showed preserved function of cortical neuronal networks in the Df(h22q11)/+ mice. Thus, expression of most receptors for the major transmitters is not altered, and the mechanism responsible for increased behavioural responsivity to NMDAr blockade is unknown, which should be investigated further in future studies.
In line with the increased ASR, the amplitude of AEPs were increased in Df(h22q11)/+ mice. The amplitude of AEPs has been found to be decreased in patients with schizophrenia,43 whereas mixed results have been found for LDAEP. Some studies have reported that LDAEP was reduced in patients with schizophrenia, but these studies were not designed to account for difference in symptomatology.44,45 In a recent study, Wyss and colleagues46 found increased LDAEP in patients with schizophrenia with predominant negative symptoms, which interestingly is also prominent in the symptomatology of patients with the 22q11.2DS47–49 together with increased auditory event-related potentials.50 Negative symptoms are also prominent in patients with autism, another disorder associated with the 22q11.2DS, and increased amplitude of AEPs have been reported in patients with autism-related syndromes, such as fragile X,51 and in animal models of autism.52,53 We have not assessed behavioural functions related to negative symptomatology in the present study. However, reports of a reduced progressive ratio response in Df(16)A+/− mice54 supports negative symptom-like behaviour in these mice.55 Hence the Df(h22q11)/+ mouse may also prove to be a useful model for aspects of the negative symptomatology in patients with psychiatric disorders.
Limitations
Genetic mouse models provide insight into the consequence of the mutation in a specific genetic and environmental context. More marked changes might have been anticipated from introducing a high-risk genetic variant comprising more than 25 genes into the mouse. However, it is important to keep in mind that the 22q11.2 microdeletion has incomplete penetrance in humans and that expression and severity of disease depends on the complete genetic makeup21 in concert with environmental influences. Thus, the Df(h22q11)/+ mouse is more appropriately thought of as a liability model rather than a disease model. This is in line with our previous findings that copy number variants conferring high risk for mental disorders significantly affect structure and function of the brain in healthy human carriers.56 In order to obtain more marked phenotypes reflecting the severe conditions related to 22q11.2DS it has been suggested to combine the Df(h22q11)/+ mouse with environmental stressors, as used by Giovanoli and colleagues,57 which may unmask latent psychopathology. Giovanoli and colleagues showed how unpredictable stress during puberty resulted in PPI deficits and increased sensitivity to amphetamine and MK-801 in mice exposed to prenatal immune activation. In addition, Harper and colleagues22 showed how reduced social interaction in Sept5 knockout mice, one of the 22q11.2 genes, could be rescued by reducing the level of stress, clearly demonstrating a genotype × environment interaction.
Sex differences in the behaviour of children with 22q11.2DS have been reported.58 Thus, it is highly relevant to explore sex differences in the mouse. In the present study only male mice were included. Pilot studies conducted in our laboratory comparing male and female Df(h22q11)/+ mice did not reveal differences in motor function, seizure threshold, anxiety, or amphetamine-induced locomotion. The studies have to be repeated and extended before firm conclusions can be made.
We observed only a few changes in protein expression. Additional synaptic fractionation might increase the resolution of the analysis, particularly with respect to synaptic proteins. However, the method used allowed us to investigate both synaptic (e.g., PSD95) and astrocytic markers (e.g., GFAP).
Conclusion
Here we show how the Df(h22q11)/+ mouse model reflects different developmental aspects of the 22q11.2DS and schizophrenia, including changes in information processing and NMDAr antagonist responsiveness. Furthermore, the present findings extends previous reports on other 22q11.2DS mouse models by showing age-independent and antipsychotic-resistant gating deficits that are not confounded by reduced hearing. This model will be a valuable tool for further understanding the etiology of schizophrenia and other psychiatric disorders with the ultimate goal of developing novel and improved drugs.
Acknowledgements
The research leading to these results was conducted as part of NEWMEDS and received support from the Innovative Medicine Initiative Joint Undertaking under grant agreement 115008, of which resources are composed of an EFPIA in-kind contribution and financial contribution from the European Union’s Seventh Framework Programme (FP7/2007–2013). This work was further supported by grants from the Danish Advanced Technology Foundation (File no. 001-2009-2) and by the Instituto de Salud Carlos III, Centro de Investigación Biomédica en Red de Salud Mental (CIBERSAM). We thank the technical staff for skillful assistance.
Footnotes
Competing interests: M. Didriksen, V. Nielsen, P. Kallunki, K. Christensen, T. Stensbøl, J. Egebjerg and J. Bastlund are employees and shareholders of H. Lundbeck A/S. K. Fejgin, M. Birknow, P. Larsen, J. Lauridsen and J. Nielsen are employees of H. Lundbeck A/S. M. Birknow has received personal fees from Innovation Fund Denmark. H. Grayton and F. Gastambide are employees of Eli Lilly & Co. Ltd, UK. T. Werge has received consultant and lecture fees from H. Lundbeck A/S. F. Artigas has received consultant fees from Lundbeck A/S and is member of the scientific advisory board of Neurolixis. He is also coinventor of patents on RNAi technologies in collaboration with nLife Therapeutics. No other competing interests declared.
Contributors: M. Didriksen, K. Fejgin, M. Birknow, J. Lauridsen, V. Nielsen, P. Kallunki, K/ Christensen, T. Werge, T. Stensbøl, J. Egebjerg, F. Gastambide, F. Artigas, J. Bastlund and J. Nielsen designed the study. M. Didriksen, K. Fejgin, M. Birknow, H. Grayton, P. Larsen, J. Lauridsen, V. Nielsen, P. Celada, N. Santana, P. Kallunki, K. Christensen, F. Gastambide and J. Bastlund acquired and analyzed the data, which S. Nilsson, F. Artigas and J. Nielsen also analyzed. M. Didriksen, S. Nilsson, M. Birknow, H. Grayton, P. Larsen, J. Lauridsen, V. Nielsen, P. Celada, N. Santana, P. Kallunki, T. Werge, F. Gastambide, F. Artigas, J. Bastlund and J. Nielsen wrote the article, which all authors reviewed and approved for publication.
- Received January 6, 2016.
- Revision received March 5, 2016.
- Accepted April 5, 2016.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools



Related Articles
Cited By...
- Early visual processing as a marker of disease, not vulnerability: Event-related potential (ERP) evidence from 22q11.2 deletion syndrome, a population at high risk for schizophrenia
- Behavioural and molecular characterisation of the Dlg2 haploinsufficiency rat model of genetic risk for psychiatric disorder
- Zebrafish as a tool to study schizophrenia-associated copy number variants
- Hearing loss promotes schizophrenia-relevant brain and behavioral abnormalities in a mouse model of human 22q11.2 Deletion Syndrome
- Disruption of the Blood-Brain Barrier in 22q11.2 Deletion Syndrome
- Analysis of genes within the schizophrenia-linked 22q11.2 deletion identifies interaction of night owl/LZTR1 and NF1 in GABAergic sleep control
- Assessing auditory processing endophenotypes associated with Schizophrenia in individuals with 22q11.2 Deletion Syndrome
- Transcriptomic networks implicate neuronal energetic abnormalities in three mouse models harboring autism and schizophrenia-associated mutations