

Abstract
Background: The heterogeneity of Alzheimer disease requires the development of multitarget drugs for treating the symptoms of the disease and its progression. Both cholinergic and monoamine oxidase dysfunctions are involved in the pathological process. Thus, we hypothesized that the development of therapies focused on these targets might be effective. We have developed and assessed a new product, coded ASS234, a multipotent acetyl and butyrylcholinesterase/monoamine oxidase A–B inhibitor with a potent inhibitory effect on amyloid-β aggregation as well as antioxidant and antiapoptotic properties. But there is a need to reliably correlate in vitro and in vivo drug release data.
Methods: We examined the effect of ASS234 on cognition in healthy adult C57BL/6J mice in a model of scopolamine-induced cognitive impairment that often accompanies normal and pathological aging. Also, in a characterized transgenic APPswe/PS1ΔE9 mouse model of Alzheimer disease, we examined the effects of short-term ASS234 treatment on plaque deposition and gliosis using immunohistochemistry. Toxicology of ASS234 was assessed using a quantitative high-throughput in vitro cytotoxicity screening assay following the MTT assay method in HepG2 liver cells.
Results: In vivo, ASS234 significantly decreased scopolamine-induced learning deficits in C57BL/6J mice. Also, reduction of amyloid plaque burden and gliosis in the cortex and hippocampus was assessed. In vitro, ASS234 exhibited lesser toxicity than donepezil and tacrine.
Limitations: The study was conducted in male mice only. Although the Alzheimer disease model does not recapitulate all features of the human disease, it exhibits progressive monoaminergic neurodegeneration.
Conclusion: ASS234 is a promising alternative drug of choice to treat the cognitive decline and neurodegeneration underlying Alzheimer disease.
Introduction
Numerous studies have focused on the pathogenic mechanism of Alzheimer disease, but our understanding of its pathophysiology is rather limited. Intraneuronal neurofibrillary tangles (NFTs), together with accumulation of amyloid β peptide (senile plaques, Aβ), constitute the major neuropathological hallmarks of Alzheimer disease.1 Although it is not clear whether abnormal processing of the amyloid precursor protein (APP) is the initial cause or rather a late event in the pathophysiology of Alzheimer disease,2 the generation of Aβ from its precursor protein APP induces oxidative stress and plays a critical role in the pathogenesis and advancement of the disease, producing neuronal injury and loss, inflammation and characteristic activation of microglia and astrocytic cells.3–6 Together with aberrant Aβ deposits in the neuropil, a genetic basis is essential in influencing the onset and/or modifying the progression of the disease.7 Genomic factors induced by environmental elements and epigenetic phenomena might be responsible for Alzheimer disease pathogenesis leading to premature neuronal death.8 For unknown reasons, accumulation of insoluble fibrous material progresses from the entorhinal cortex toward regions involved in cognitive functions, particularly learning and memory.9,10
In addition to a cholinergic deficit, which correlates with the severity of cognitive symptoms,10 neuropsychological symptoms, such as depression and anxiety, often observed together with cognitive deficits in patients with Alzheimer disease, suggest that dysfunction of the dopaminergic systems might be further implicated.9,11 Presently, curing the disease continues to be out of reach, and stimulating cholinergic neurotransmission remains the only successful approach that improves the cognitive state. In this regard, cholinesterase enzymes, such as acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE), are the main targets for a therapeutic approach. Over the years, many different ChE inhibitors (ChEIs) have been developed to boost the central cholinergic transmission, but approved ChEI-based treatments have proven to provide only transitory and modest recovery in cognitive impairment without affecting the natural progression or the ultimate outcome of the disease.12
Multitarget disease-modifying drugs, in which a single molecule is directed to bind multiple sites, have been designed as drug candidates with potential to halt the progression of the disease.13 The pivotal role of Aβ in the pathogenesis of Alzheimer disease and the proven cognitive benefit of cholinergic system augmentation support the development of multitarget molecules targeting both the cholinergic system and Aβ deposits. Activated monoamine oxidase-B (MAO-B) in the brains of patients with Alzheimer disease has been shown14 to contribute to the production of free radicals and oxidative stress observed in the pathogenesis of Alzheimer disease. In addition to the degeneration of cortical and hippocampal neurons, Alzheimer disease is associated with the early and progressive degeneration of monoaminergic neurons.15,16 For these reasons, the development of hybrid drugs that behave as dual inhibitors of both ChEs and MAO are being actively produced to treat Alzheimer disease symptoms and potentially slow the disease progression.17–19 In this regard, we have conceived a compound coded ASS234 (see the structure in the Methods section),20–22 that is a a multipotent AChE/MAO inhibitor with the ability to cross the blood–brain barrier and that shows antioxidant and neuroprotective properties with inhibitory effects on Aβ aggregation.23,24 Beneficial effects of ASS234 have been shown using the hole-board test.18 To correlate in vitro and in vivo drug release data, we focused on the evaluation of ASS234 as a cognitive enhancer in a scopolamine-induced cognitive impairment model in C57BL/6J mice. In addition, we tested the therapeutic potential of ASS234 to dissolve and prevent Aβ aggregate formation in a transgenic mouse model of Alzheimer disease, APPswe/PS1ΔE9,25 that exhibited amyloid pathology associated with progressive monoaminergic neurodegeneration.26,27 We hypothesized that ASS234 would be a promising cognitive-enhancing drug that would reduce amyloid plaque deposition.
Methods
Chemicals
A new MTDL compound ASS234 (benzylpiperidine-indolyl-proparglamine molecule; Fig. 1) was synthesized, as described previously.22

New MTDL compound ASS234 (benzylpiperidine-indolyl-proparglamine molecule).
In vivo experiments
All procedures with animals were carried out in accordance with European Communities Council Directive (2010/63/UE) on animal experiments under a protocol approved by the Animal Welfare Committee of the Cajal Institute (Madrid, Spain) and by the Institutional Animal Ethics Committee of the Spain Council for Scientific Research (CSIC), adhering to the recommendations of the European Council and Spanish Department of Health for Laboratory Animals (R.D. 53/2013). A special effort was made to reduce the number of animals used in the study, and the number of animals assigned to each group was to be kept to a minimum necessary to achieve adequate significance.
Behavioural experiments
For behavioural studies, we used 60 male 8-week-old C57BL/6J mice (Harlan) weighing 25–30 g.
Memory impairment was induced by the potent antimuscarinic drug scopolamine hydrobromide (0.5 mg/kg, Sigma) dissolved in saline buffer. To explore the potential of ASS234 as a cognitive enhancer, we administered the hybrid drug at a dose of 0.62 mg/kg (0.12 mM/kg). We tested the antiamnesic effect of donepezil (Tocris Bioscience, R&D Systems Inc.) at a dose of 0.12 mM/kg. Both products, ASS234 and donepezil, were dissolved in saline buffer containing 3.6% dimethyl sulfoxide (DMSO; AppliChem). All drug solutions were independently prepared immediately before their use and were administered intraperitoneally in 10 mL/kg of vehicle volume. Different batches of mice were used for each experiment. The animals were distributed into 6 experimental groups (I–VI): control groups (I, II) consisted of (I) untreated mice, labelled as “control nontreated” and (II) mice administered 3.6% DMSO in saline buffer, labelled as “control vehicle”; experimental groups (III–V) consisted of (III) mice administered scopolamine dissolved in 3.6% DMSO in saline buffer, labelled as “scop/DMSO,” (IV) mice coadministered scopolamine plus ASS234, labelled as “ASS234/scop,” and (V) mice coadministrated scopolamine plus donepezil, labelled as “done/scop.” The combined treatments in groups III–V were given simultaneously, one by one by using separate sterile needles (25G) and syringe for each drug. Group VI (n = 6) was conceived to explore the possibility that treatment with ASS234 might have some effect on naive C57BL/6 mice.
Object recognition task (ORT)
Spontaneous object recognition was tested as described previously,28 with slight modifications. All behavioural experiments took place during the light phase of the 12-h light/dark cycle between 10 am and 3 pm to minimize the influence of circadian rhythm, and they were conducted with the investigators blind to treatments. Prior to the experiment, mice were handled individually for several days. The ORT was performed in a Plexiglas open field square box (40 × 40 × 40 cm) with black walls and the floor covered with sawdust. The apparatus was placed in a poorly illuminated (60 lux) and noise-isolated room, and the mice were randomly placed in the experimental room 30 min before beginning the experimental task each day. The behaviour of the animals (time spent exploring the objects and overall locomotor activity) was registered during precise time periods. Briefly, during the 3 consecutive days preceding the test day, mice were habituated to an empty open field box. On the first day animals were allowed to explore the apparatus individually for 5 min in the absence of objects (habituation session). On the second and third days animals were allowed again to explore the apparatus for 10 min in the absence of objects. On the test day, 30 min before the trial session, mice were administered treatments as indicated in the previous section and then subjected to the acquisition trial (T1) and the retention trial (T2). In T1 mice were individually allowed for 6 min to freely explore 2 identical plastic objects placed in the arena, located in symmetric positions at a distance of about 7 cm away from walls. Then the rodents were removed from the environment for an intertrial interval of 1 hr. After the delay period, animals were returned for 5 min (T2) to the bin where 1 of the original objects had been replaced by a novel object (neutral to naive mice) with a different shape, colour and textures than the remaining object previously presented in T1 (familiar object). To score the exploration behaviour of the mice automatically, a camera (Panasonic VDR-D 150E) was mounted above the centre of the arena. The total time spent in active exploration of each object during T1 and T2 was determined with a computerized tracking system and image analyzer (Ethovision XT, Noldus Information Technology) that tracked the nose, centre and tail points of each mouse.29 As a criterion of active object exploration, we measured the time spent by the animal directing its nose to the object at a distance of less than 2 cm and/or touching it with the vibrissae or nose. Mice exploring the objects for less than 15 s within the 6-min period of T1 were discarded from experiments; only mice having a minimal level of total object exploration of 15 s (novel object plus familiar object ≥ 15 s) during T2 were included in the study. Recognition memory differences between the familiar and the new objects is operationally defined as a recognition index (RI) that corresponds to the proportion (%) of time spent exploring the novel object with respect to total time spent exploring both the novel and familiar objects: RI = tnovel ÷ (tnovel + tfamiliar) × 100. To exclude spatial preferences, mice showing no preference for either object during T1 (RI = 50%) were used in this study.
Studies on Aβ load and gliosis
We tested the efficacy of ASS234 in lowering Aβ plaque load and gliosis in vivo using immunohistochemistry (IHC). Experiments were performed on 10 week-old male APPswe/PS1ΔE9 transgenic mice (n = 14) that were either vehicle control or treated with ASS234 for 16 weeks. A group of mice (n = 8) were treated daily with ASS234 at a dose of 0.62 mg/kg/d by using ALZET mini-osmotic pumps (ALZET, model 2004) that were subcutaneously implanted. Pumps were replaced each 28 days. The control group (n = 6) was treated with vehicle: saline buffer containing 3.6% DMSO. Mini-osmotic pumps are widely used for the study of neurodegenerative diseases. The dose of ASS234 used in this study has proven effective cognitive enhancing action in the object recognition test (see the Results section). In all cases, treatment was initiated at the age of 10 weeks, as no plaques or very few can be detected at that age. After 4 months of treatment, subsequent histological studies were performed following the histochemical procedures described in the sections that follow. During treatment, body weight was recorded once a week. We statistically analyzed the differences between the groups.
Amyloid plaque localization and gliosis
Immunohistochemistry
Experimental animals were processed according to standard procedures already published by our group.30 Briefly, at the end of the treatment, the brains were fixed by transcardial perfusion with 100 mL 4% (w/v) paraformaldehyde in 0.1 M phosphate buffer (PB), pH 7.4. Afterwards, brains were removed, cut into blocks and fixed in the same fixative for 4 h at room temperature. Brains were then cryopreserved in 30% sucrose in 0.1M phosphate buffer pH 7.4 overnight at 4ºC. Then, 30 mm thick coronal frozen sections were obtained with the aid of a cryostat (Leica CM 1900). Sections were stored at −20°C in a cryoprotectant solution (30% glycerol and 30% ethylene glycol in 0.1 M phosphate buffered saline) until use. Selected free-floating sections were processed using the avidin–biotin peroxidase complex (ABC) technique to visualize endogenous Aβ with rabbit β-amyloid antibody (Cell Signalling 1:500), microglia with polyclonal anti-Iba-1 antibody (Wako 1:500), and glial fibrillary acidic protein (GFAP) with polyclonal anti-GFAP antibody (DAKO 1:1500). Anti-rabbit biotinylated secondary antibodies and peroxidase-linked ABC kit were purchased from Vector laboratories. Light microscopy photomicrographs were taken with a digital camera attached to a bright-field microscope. In addition to light microscopy IHC, Aβ deposits were also visualized using the thioflavin S staining procedure31 with a fluorescence microscope.
Quantification of amyloid plaque load and gliosis
Following 4 months of treatment, mice were sacrificed to proceed with the statistical evaluation of the increase in the number of Aβ plaques and gliosis generated, both in the cerebral cortex and hippocampus. A selection of sections from experimental animals, obtained according to Paxinos and Franklin32 between Bregma levels AP −1.3 mm and −2.20 mm, were processed (10 sections per case) and then photographed. For statistical analysis, amyloid plaques stained by IHC or thioflavin S were photographed at a very low magnification with the aid of a Nikon Eclipse E400 microscope or a LEICA AF 6500–7000 microscope, respectively. The number of identified Aβ plaques in microscopy images was obtained using NIS-Elements BR 3.2 Nikon software for IHC or the ImageJ software for thioflavin S staining. The number of cells positive for GFAP or Iba-1 in the vicinity of neuritic plaques was also counted from microscope images taken from histological sections obtained at the same AP levels as those for Aβ plaque detection (10 sections per case). Images were taken at higher power (×20) with a Nikon Eclipse 80i microscope connected to a workstation using Stereo Investigator software. At this magnification, stained cell bodies were easily identified. To avoid double counting, cells were counted when their nuclei were optimally visualized, which occurred usually in a single focal plane.
In vitro experiments
Culture of HepG2 liver cells and treatment
The human hepatoma cell line HepG2 was kindly provided by IdiPAZ Institute for Health Research (Madrid, Spain). The cells were cultured in Eagle minimum essential medium (EMEM) supplemented with 15 nonessential amino acids, 1 mM sodium pyruvate, 10% heat-inactivated fetal bovine serum (FBS), 100 units/mL penicillin, and 100 μg/mL streptomycin (reagents from Invitrogen). Cultures were seeded into flasks containing supplemented medium and maintained at 37ºC in a humidified atmosphere of 5% CO2 and 95% air. For assays, HepG2 cells were subcultured in 96-well plates at a seeding density of 8 × 104 cells per well. When the HepG2 cells reached 80% confluency, the medium was replaced with fresh medium containing 1–1000 μM of ASS234, tacrine and donepezil, with 0.1% DMSO as a vehicle control (Table 1).
In vitro viability (%) of HepG2 liver cells by compounds ASS234, donepezil and tacrine*
MTT assay and cell viability
Cell viability, via the mitochondrial activity of living cells, was measured using quantitative colorimetric assay with 3-[4,5 dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT; Sigma Aldrich), as described previously.33 We analyzed triplicate samples of at least 4 different cultures.
Statistical analysis
Mouse behaviour was analyzed by applying a 1-way analysis of variance (ANOVA), followed by a Bonferroni post hoc test for multigroup comparisons to compare the data from the experimental groups. In relation to quantitative assessment of amyloid plaques and gliosis, statistical differences between 2 sets of groups were determined using an unpaired t test. Comparisons between drugs and control groups in cell viability were performed using 1-way ANOVA, followed by a Newman–Keuls post hoc test. Results are expressed as means ± standard errors of the mean, and we considered differences to be significant at p < 0.05.
Results
Robust enhancing effect of single-dose ASS234 on cognition
The effects of drug treatment on mouse behaviour are represented in Figure 2. As expected, scopolamine significantly reduced the exploratory preference for a novel object in T2. Scopolamine-induced cognitive deficit was reflected by decreased RI in comparison with nontreated and vehicle control groups (p = 0.009 and p = 0.049, respectively; Fig. 2A). In striking contrast, significant increase of the RI, indicating reversal of memory impairment induced by scopolamine was obtained with a single dose of donepezil or ASS234 (p = 0.049 and p = 0.027, respectively). As shown in Figure 2A, the scopolamine-induced amnesia was consistently reversed by concomitant administration of donepezil (0.12 mM/kg), which significantly improved the cognitive performance by about 13.1% (p = 0.049) compared with the scop/DMSO group. Administration of ASS234 had similar cognitive effects as donepezil at the same dose range (14.1% improvement compared with the scop/DMSO group, p = 0.027; Fig. 2A), suggesting that ASS234 exerts its therapeutic effect by enhancing natural memory processes. No differences were observed between groups II and VI (data not shown).
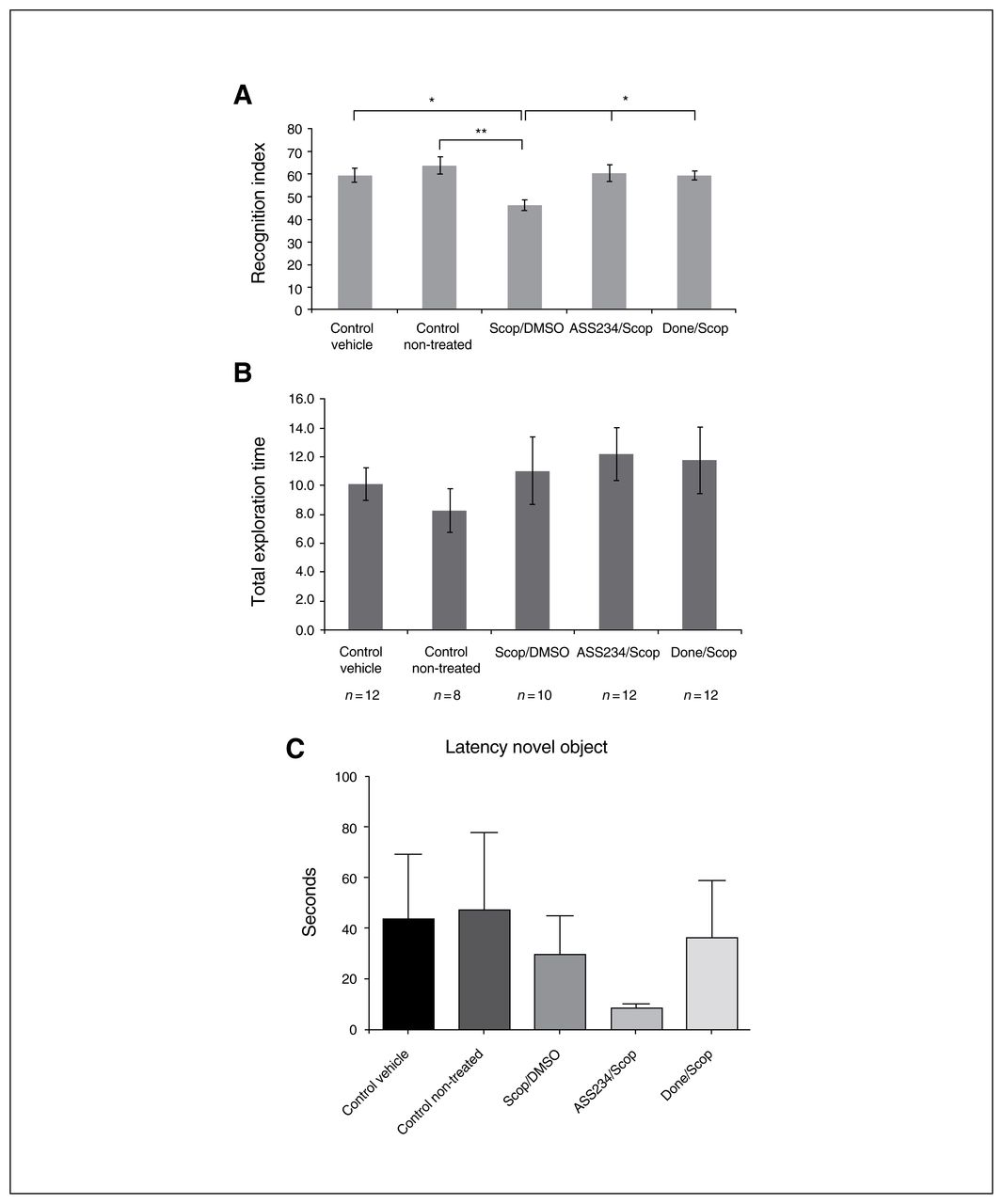
Effect of combined administration of scopolamine/dimethyl sulfoxide (scop/DMSO), donepezil and ASS234 on (A) recognition index (RI) and (B) total objects exploration time in T2. Drugs were coadministered 30 min before T1. (A) Significant differences between scop/DMSO and control nontreated groups (**p < 0.01). When compared with the scop/DMSO group, treatment with either donepezil (done/scop group) or ASS234 (ASS234/scop group) (0.12 mM/kg) reversed the short-term memory impairment induced by scopolamine (*p < 0.05), suggesting that ASS234 is able to reverse scopolamine-induced cognitive impairment. The dose chosen for the hybrid drug (0,12 mM/kg) was the same as that of donepezil (0.12 mM/kg), for comparative purposes. The percentage of the novel object recognition time was evaluated as RI = (tnovel ÷ [tnovel + tfamiliar]) × 100. (B) No significant effect on total exploration time during the test was found after intraperitoneal administration of scop/DMSO, donepezil or ASS234. (C) There were no differences between groups in the latency to first approach to the novel object. Behavioural data were analyzed using 1-way analysis of variance (ANOVA), followed by a Bonferroni post hoc test for multigroup comparisons to compare the data from the experimental groups.
In striking contrast, we found no differences in the total exploration time between the different experimental groups (Fig. 2B), which correlates well with the fact that donepezil leads to no difference between treatment conditions in the level of exploration in T1 or T2.34 As expected, no significant effect on total object exploration time during the test was found after intraperitoneal administration of ASS234 or donepezil (data not shown). Also, we observed no hyper- or hypoactivity following ASS234 or donepezil administration. Characteristically, the different groups of mice did not differ in the latency to first approach the novel object (Fig. 2C).
Amyloid plaque burden and gliosis were decreased in the cortex of the ASS234-treated group of APPswe/PS1ΔE9 transgenic mice
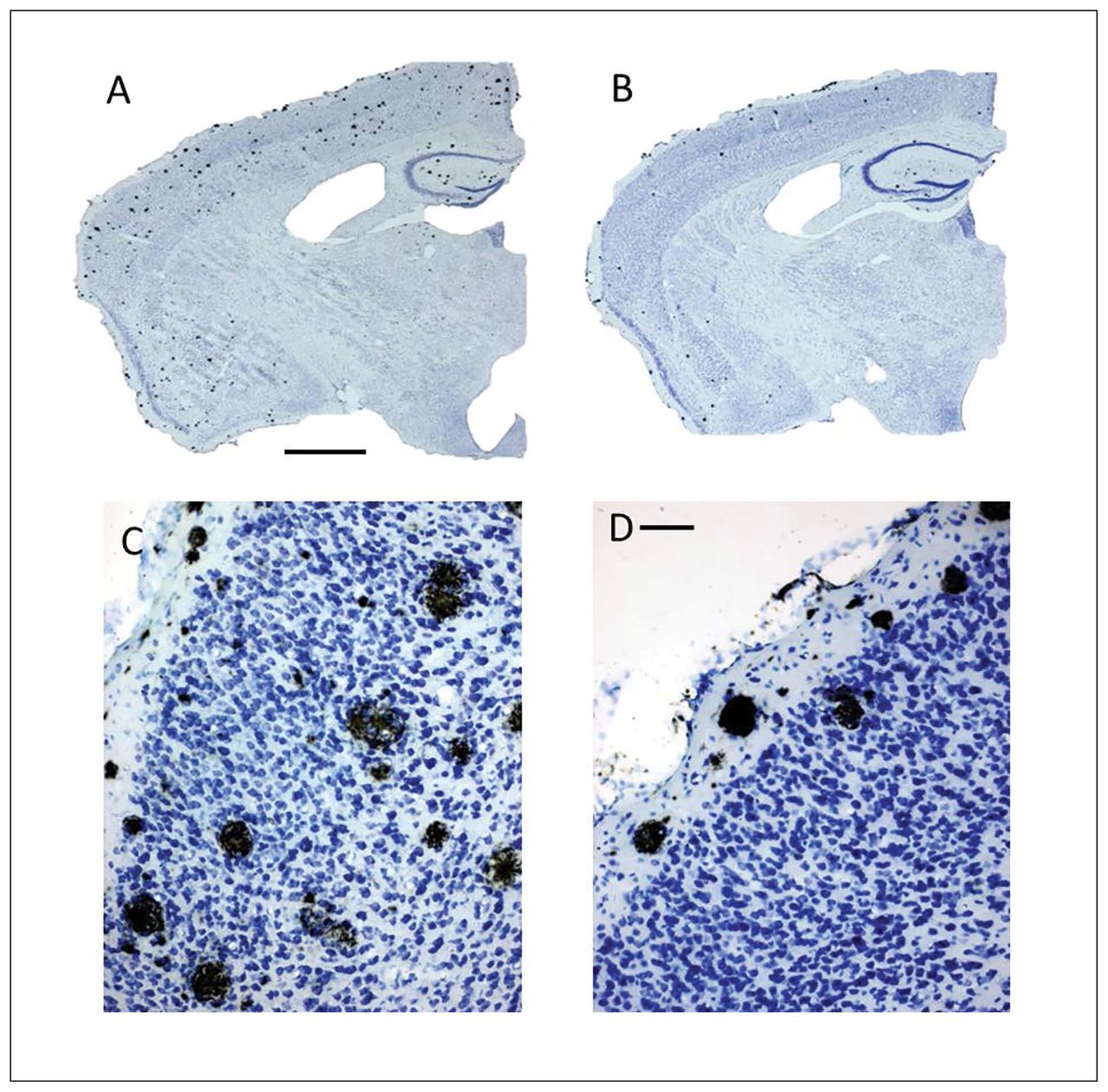
To determine the effect of ASS234 on Aβ plaque burden at the end of the treatment, we evaluated the location and number of neuritic plaques throughout the cortex and hippocampus using IHC and thioflavin S staining procedures. Characteristically, body weight did not differ between the groups after 4 months of treatment (data not shown). With IHC, we observed an apparent reduction in the number of neuritic plaques in the cerebral cortex and hippocampus in ASS234-treated transgenic mice compared with vehicle-treated mice (Fig. 3). Further statistical analysis confirmed the IHC results (Fig. 4). Cortical plaque deposition was significantly decreased (unpaired t test, p = 0.002) in ASS234-treated transgenic mice compared with controls (Fig. 4A). As in the cortex, Aβ plaque load in ASS234-treated transgenic mice showed a trend toward a reduction in the hippocampus (Fig. 4B). These findings indicate that ASS234 has a greater effect on reducing plaque load in cerebral cortex than in hippocampus of APPswe/PS1ΔE9 transgenic mice.

Coronal brain sections processed for single detection of Aβ peptide using immunohistochemistry (IHC) counterstained with Nissl stain. The sections shown are between Bregma levels −1.50 mm and −1.80 mm.32 (C–D) High-power magnifications of the same transgenic mice shown in panels A and B, respectively. (B, D) Transgenic mice treated with ASS234 apparently show a much lower number of Aβ plaques in the cortex than (A, C) vehicle-treated control mice. In panels B and D, neuritic plaques are mainly accumulated in superficial layers of the cortex. Scale bars: A and B: 500 μm; C and D: 50 μm.
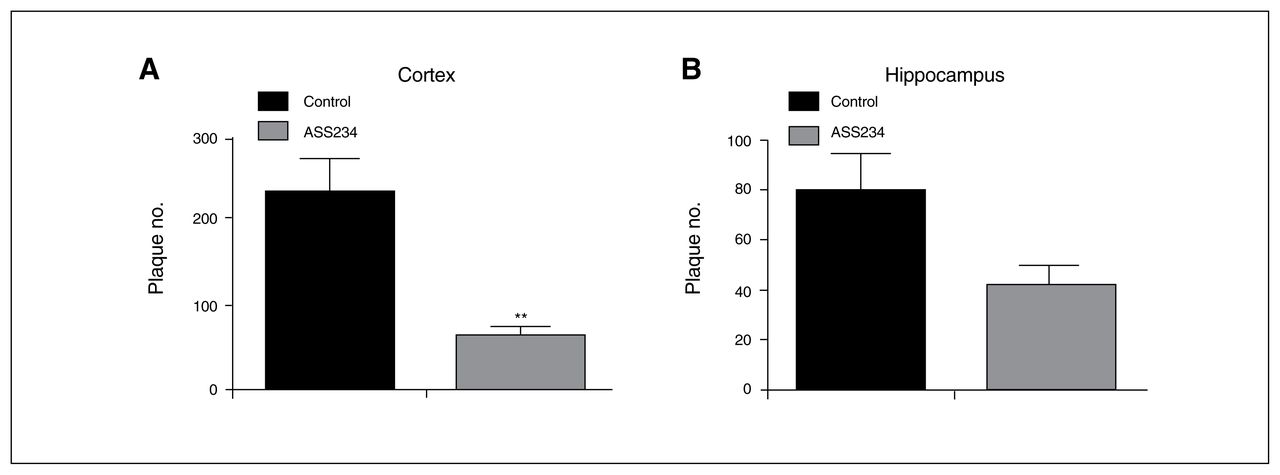
Mean values of plaque load in the cerebral cortex and hippocampus taken from 40 μm-thick coronal sections stained to detect endogenous Aβ by immunohistochemistry. (A) Statistical evaluation revealed a significant decrease in plaque load in the cortex of transgenic mice treated with ASS234 compared with transgenic vehicle-treated mice (unpaired t test, p = 0.002). (B) A trend toward a reduction in plaque load was observed in the hippocampus. Data are represented as means ± standard errors of the mean.
As microgliosis and astrocytosis are prominent aspects of this Alzheimer disease mouse model indicative of neuroinflammation,35–38 we identified the effect of ASS234 on neuroinflammation through the evaluation of the immunohistochemical distribution of the astrocyte marker protein GFAP (Fig. 5A, B and E) and of the microglia/macrophage-specific protein Iba1 (Fig. 5C, D and F). Significantly reduced GFAP and Iba1 immunostainings (Fig. 5E and F, respectively) were observed in the cortex of the ASS234-treated transgenic mice compared with that of controls (unpaired t test, p < 0.001), suggesting a beneficial effect of ASS234 on neuroinflammation.

Light microscopy immunohistochemistry showing the cellular location of immunoreactive sites for (A–B) endogenous GFAP and (C–D) Iba1 in the cortex of (A and C) vehicle- and (B and D) ASS234-treated transgenic mice. In panels A and B labelled astrocytes surround neuritic plaques, and the number of immunoreactive elements in panel A appears to be much higher than in panel B. When comparing panels C and D, Iba1 immunoreaction product in panel C labels microglial cells with large densely stained thick processes, suggesting much higher microglial activation in vehicle-treated transgenic mice. (A–D) Scale bar: 30 μm. (E–F) Mean values of the number of GFAP- and Iba1-stained cells in the cortex of ASS234- and vehicle (control)–treated transgenic mice, respectively. Statistical evaluation revealed a significant decrease in the number of GFAP- and Iba1-stained cells in transgenic mice treated with ASS234 than in vehicle-treated mice (unpaired t test, p < 0.001). This reduction was not statistically detected in the hippocampus (data not shown). Data are represented as means ± standard errors of the mean. GFAP = glial fibrillary acidic protein.
In vitro experiments
The promising results shown above and the fact that anti-dementive drugs produce side effects, including gastrointestinal and hepatic alterations, led us to assess the hepatotoxicity of compound ASS234. As an example, tacrine treatment for Alzheimer disease that was approved by the US Food and Drug Administration in 1993 was discontinued owing to the hepatotoxicity seen in about 30%–50% of the treated patients.39 Very recently, Chew and colleagues40 showed that donepezil was able to induce hepatotoxicity. Toxicity studies of ASS234 along with donepezil and tacrine were performed in parallel in the human cell line HepG2. The results showed that the 3 compounds reduced cell viability in a concentration-dependent manner (Table 1). However, at very high concentrations (100 μM and 300 μM) ASS234 exhibited less toxicity than the reference compounds donepezil and tacrine.
Discussion
Because cholinergic deficit is a consistent and early finding in patients with Alzheimer disease, AChE remains a highly viable target for the symptomatic improvement in these patients.17 The multifactorial nature of Alzheimer disease indicates that use of a ChE inhibitor or MAO inhibitor alone does not significantly delay or halt disease progression. This is the reason why synthesis of compounds capable of targeting different enzymatic systems related to Alzheimer disease (ChEs and MAO-A/B) are actively pursued,41 and synthesis of donepezil-related compounds possessing metal-chelating properties is strongly supported for the treatment of Alzheimer disease.42 In a first part of our study we examined the effect of ASS234 on cognition in healthy adult C57BL/6J mice in a model of scopolamine forgetting. This study confirms in vivo cholinergic properties of ASS234 to be as efficacious in cognition as treatment with donepezil. It is noteworthy that clinical studies revealed that AChEIs have a small beneficial effect on amyloid plaque load. We have shown in a previous study that the potent inhibitory AChE profile of ASS234 and the presence of the propargylamine group of the MAO inhibitor reinforces its action against the aggregation of the most physiologically relevant β-amyloid isoforms.23 Since mechanisms for memory loss are related to the amount of soluble Aβ oligomers43,44 and MAO-B is overexpressed in the brains of patients with Alzheimer disease,14 compounds showing dual AChE and MAO inhibition with antioxidant and inhibitory effects on Aβ-aggregation are potential candidates for treating the progression of Alzheimer disease.45 Drugs currently developed are site-activated metal-chelators able to inhibit both AChE and MAO–A/B,17 but this class of medications has not been established to possess disease-modifying properties or reduction of dose-related side effects of AChEIs based on MAO inhibition.
Our group has investigated the effect of the introduction of a benzyloxy group in a series of acetylenic and allenic derivatives of tryptamine that were previously reported to be potent and selective inhibitors for MAO-B.46 On the basis of this previous work, we have designed novel compounds to act as dual inhibitors of MAO and AChE, such as the hit compound ASS234. In vitro, the multitarget compound ASS234 retains activity against human AChE and BuChE as well as MAO-A/B.22–24,47,48 Through inhibition of Aβ1–42 and Aβ1–40 self-aggregation, thus limiting fibrillary and oligomeric species formation, ASS234 proved to be neuroprotective against Aβ toxicity.23,49 We have shown in vitro that ASS234 may have an impact on different processes involved in Alzheimer disease pathogenesis.22–24 Whereas our present results do not allow us to provide data on the molecular mechanisms underlying the neuroprotective effects of ASS234, we have recently proven in vitro activation of Wnt signalling by ASS234.49 Potentiation of Wnt signalling rescues memory loss and improves synaptic dysfunction in animal models of Alzheimer disease,50 suggesting that the Wnt signalling pathway might represent a pharmacological target for the neuroprotective effects of this drug in APPswe/PS1ΔE9 mice. The possibility that ASS234 can prevent Aβ-induced GSK3b activation in APPswe/PS1ΔE9 mice should be considered in future research.
Based on these previous findings and the fact that ASS234 crosses the blood–brain barrier,18 we carried out an in vivo study to get the proof-of-concept of this multitarget directed ligand for Alzheimer disease.
The present research discusses the effect of an AChE-BuChE/MAO-A/B inhibitor, ASS234, and the AChE inhibitor donepezil on acute scopolamine-induced cognitive deficit in ORT using wild type C57BL/6 inbred mouse strain. The novel object recognition paradigms in animals constitute a valuable measure of cognition.51 Although scopolamine-induced deficits in the cholinergic system produce a memory acquisition deficit that does not replicate or model Alzheimer disease–like pathology, these deficits do make an appropriate model for evaluating short-term memory and in vivo effects of AChEIs. Our results confirm cholinergic properties of ASS234 to be as efficacious as treatment with donepezil. But the proposed therapeutic approach is still incomplete and does not provide data regarding prevention of inevitable neuronal degeneration. Thus, we assessed the effect of ASS234 on amyloid plaque burden in a fully characterized animal model of Alzheimer disease useful for preclinical studies.27,35,36,38 The recapitulation of amyloid pathology and cognitive deficits as seen in patients with Alzheimer disease and the relative quick onset of pathology and symptoms makes the APPswe/PS1ΔE9 transgenic mouse model suitable for investigating Alzheimer disease–related processes. Our results — that ASS234 treatment alleviates scopolamine-induced disruption of acquisition memory and improves Alzheimer disease pathology in mice overexpressing APP — provide in vivo data that suggest ASS234 may limit Alzheimer disease progression.
The APPswe/PS1ΔE9 transgenic mice used in the present study display amyloid deposition at a very early age (10–12 wk). Histopathologically, this model is characterized by the presence of extracellular senile plaques consisting of Aβ peptide in the cortex, hippocampus and other regions of the brain associated with progressive monoaminergic neurodegeneration.26 Treatment with ASS234 reduced the number of neuritic plaques in the cerebral cortex and resulted in a trend toward a reduction in the hippocampus, as seen with IHC and thioflavin S staining, the most widely used “gold standards” for selectively staining and identifying amyloid fibrils in vivo.31 These differences might be associated with age and duration of treatment, as previously reported.52
According to the amyloid cascade hypothesis, Aβ peptide accumulates into plaques, triggering a series of events leading to dementia.1 However, this theory fails to explain the discrepancy between amyloid plaque deposition and cognitive impairment in patients with Alzheimer disease.5 Clinico-pathological correlation studies and positron emission tomography amyloid imaging studies have shown that some individuals can tolerate substantial amounts of Alzheimer disease pathology in their brains without experiencing dementia.5 Although the significance of plaque load for Alzheimer disease is still under debate and several groups found the pattern of amyloid plaques to be of limited relevance for the neuropathological staging of Alzheimer disease, results from other laboratories showed a strong correlation between counts of senile plaques in the hippocampus and cortex and memory function. Glial activation emerged as a likely mediator of neurotoxicity and altered cognition, providing further insight into factors and pathways potentially involved in human susceptibility or resilience to Alzheimer disease.5 Characteristically, patients with dementia exhibit robust glial activation, which is greatly reduced in individuals without dementia. Microglial activation parallels Aβ plaque burden in patients with Alzheimer disease and is thus a characteristic feature of Alzheimer disease progression.53 Untreated transgenic animals exhibited densely Iba1-stained microglial cells with thick processes as reported in transgenic animals.54 The fact that the size and number of microglial cells are greater in untreated transgenic mice may underlie that treatment with ASS234 might prevent microglial activation. Beyond microgliosis, reactive astrocytes are the second major component of neuroinflammation in patients with Alzheimer disease. In response to damage, astrocytes change from their normal quiescent state into a reactive state. This process, called reactive gliosis, is characterized by a profound increase in the expression of GFAP.55 In the APPswe/PS1ΔE9 transgenic mice, astrocytosis develops in parallel with plaque deposition, with severe gliosis around 6 months, especially in the vicinity of plaques. Characteristically, the number of GFAP-positive cells progressively increases with age in the APPswe/PS1ΔE9 transgenic mice.36 As in the brains of patients with Alzheimer disease, neuroinflammation in the area of amyloid plaques was observed in mouse models of Alzheimer disease, including the APPswe/PS1ΔE9 transgenic mice, as shown by astrocytic and microglial reactivity (gliosis)35,36,38 and association between neuroinflammation markers and soluble or insoluble Aβ or fibrillar amyloid plaque loads.56 To identify the effect of ASS234 treatment on neuroinflammation, we thus examined whether anomalous astrocytic and microglial activation in transgenic mice was reversed by ASS234 treatment. We observed a significant reduction in reactive gliosis after ASS234 treatment.
Limitations
Although no animal model recapitulates all features of the human disease, the APPswe/PS1ΔE9 transgenic mouse model exhibits progressive monoaminergic neurodegeneration and would be the best model to study ASS234 drug effects. Aβ deposition has age-dependent effects on cholinergic and monoaminergic fibre networks in our animal model.26,57 Selective loss of monoaminergic neurons is evident in aged animals, starting at age 18 months.26 In striking contrast, the cholinergic neuron number remains unchanged,57 Thus, the APPswe/PS1ΔE9 transgenic mouse model failed to show significant cholinergic/monoaminergic degeneration or learning and memory deficits during our study. Given this inherent weakness in the APPswe/PS1ΔE9 Alzheimer disease model, the effect of ASS234 was related only to amyloid plaque load. Given the associations between amyloid plaques, neuronal loss and dementia, our study analyzed the influence of ASS234 on Aβ deposition in the brain parenchyma as an appraisal for the development of therapeutics. Evaluation of gliosis was possible only with IHC. As in the cerebral cortex, reduction in plaque load was observed in the hippocampus, although such a reduction did not reach significance. This might be related to age and duration of treatment, as previously reported.52
Conclusion
Both the strong correlation of neuroinflammation with amyloid burden56 and our results showing that ASS234 reduces both amyloid burden and inflammation in the cerebral cortex of treated transgenic mice may underlie the beneficial action of ASS234 in slowing the progression of Alzheimer disease.
Acknowledgements
The authors thank Soledad Martinez Montero for excellent technical assistance. The study received research support from the MINECO (Government of Spain grant SAF2015-65586 awarded to R. Martínez-Murillo and J. Marco-Contelles and grant SAF2012-33304 awarded to J. Marco-Contelles). The authors thank Universidad Camilo José Cela for support (grant 2014-35).
Footnotes
↵* These authors contributed equally to this work.
Competing interests: None declared.
Contributors: J. Marco-Contelles, M. Unzeta and R. Martínez-Murillo designed the study. M. Serrano, R. Herrero-Labrador, H. Futch and A. Romero acquired the data, which M. Serrano, R. Herrero-Labrador, A. Romero, J. Serrano, A. Fernandez, A. Samadi, M. Unzeta and R. Martínez-Murillo analyzed. H. Futch, J. Marco-Contelles and R. Martínez-Murillo wrote the article, which all authors reviewed and approved for publication.
- Received June 2, 2015.
- Revision received January 3, 2016.
- Revision received March 10, 2016.
- Revision received March 22, 2016.
- Accepted March 28, 2016.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools



Related Articles
Cited By...
- No citing articles found.