

Abstract
Background: Effectively reducing the expression of certain aversive memories (fear or trauma memories) with extinction training is generally viewed to be therapeutically important. A deeper understanding of the biological basis for a more effective extinction process is also of high scientific importance.
Methods: Our study involved intraventricular injection or local injection into the dorsal dentate gyrus of anti-neuregulin 1 antibodies (anti-NRG1) before fear extinction training, followed by testing the expression of fear memory 24 hours afterward or 9 days later. We used local injection of chemogenetic or optogenetic viruses into the dorsal dentate gyrus to manipulate the activity of the dorsal dentate gyrus and test the expression of fear memory. We also examined the effect of deep brain stimulation in the dorsal dentate gyrus on the expression of fear memory.
Results: Mice that received intraventricular injection with anti-NRG1 antibodies exhibited lower expression of fear memory and increased density of activated excitatory neurons in the dorsal dentate gyrus. Injection of anti-NRG1 antibodies directly into the dorsal dentate gyrus also led to lower expression of fear memory and more activated neurons in the dorsal dentate gyrus. Inhibiting the activity of dorsal dentate gyrus excitatory neurons using an inhibitory designer receptor exclusively activated by designer drugs (DREADD) eliminated the effects of the anti-NRG1 antibodies. Enhancing the activity of the dorsal dentate gyrus with an excitatory DREADD or optogenetic stimulation resulted in lower expression of fear memory in mice that did not receive infusion of anti-NRG1 antibodies. Deep brain stimulation in the dorsal dentate gyrus effectively suppressed expression of fear memory, both during and after fear extinction training.
Limitations: The mechanism for the contribution of the dorsal dentate gyrus to the expression of fear memory needs further exploration.
Conclusion: Activation of the dorsal dentate gyrus may play an important role in modulating the expression of fear memory; its potential use in fear memory extinction is worthy of further exploration.
Introduction
A Pavlovian conditioning paradigm has been widely used to investigate the biological basis of fear memory and fear extinction. 1 Fear extinction in laboratory animals has also been used to model exposure therapy in humans — a treatment commonly used for anxiety disorder.2,3 Extinction of responses to fearful stimuli is important for the effective treatment of these disorders, especially in the context of psychiatric disorders such as posttraumatic stress disorder (PTSD) and specific phobias. It has been postulated that people with these disorders may exhibit stronger fear memory or an impaired ability to inhibit fear responses to conditioned stimuli, leading to poor extinction efficacy.
Neuregulin 1 (NRG1), a trophic factor, belongs to a family of growth factors and is expressed in both developing nervous systems and adult brains.4,5 Previous studies have shown that neutralizing endogenous NRG1 inhibits tonecued fear conditioning,6 and that NRG1 expression is significantly increased in the medial prefrontal cortex after tone-cued fear conditioning.7 In previous studies, elevated NRG1 signalling was found after anti-NRG1 injection and induced schizophrenia-like phenotypes independent of ErbB4 signalling.8,9 During phenotype analysis of these mice, we found reduced fear expression during subsequent recall. These results provided a potentially interesting scenario in which fear expression could be modulated, providing an opportunity to better understand the key brain regions involved in fear expression and identify possible mechanisms for more efficacious treatment.
The dentate gyrus plays an important role in learning and memory,10–13 but its effect on auditory fear conditioning is poorly understood. In a previous study, we found that the dorsal dentate gyrus (dDG) participated in auditory fear extinction, regulating fear renewal.14 After fear extinction training, the return of fear responses to a cue in a context that is different from the context used in extinction (i.e., fear renewal) is widely believed to represent limited efficacy of exposure therapy. We found that mice without significant fear renewal showed elevated levels of Dnmt3a in the dDG, and that overexpression of Dnmt3a resulted in a higher density of c-Fos neurons in the dDG. We also found that overexpression and knockdown of Dnmt3a in the dDG could regulate fear renewal in a bidirectional manner. The findings of that study suggested that elevated activity in the dDG may be associated with reduced fear expression.
In the present study, we used a combination of behavioural analysis, immunocytochemistry and chemogenetic manipulations to show that elevated activation of dDG excitatory neurons after infusion of anti-NRG1 antibodies into the brain led to reduced levels of freezing. To confirm this result, we selectively activated dDG excitatory neurons by expressing calcium/calmodulin-dependent protein kinase II α (CaMKIIα)–human muscarinic-3 receptor designer receptor exclusively activated by designer drugs (DREADD)-mutation (hM3D) or CaMKIIα–H134R mutation in channelrhodopsin-2 (hChR2) in the dDG and evaluated changes in fear expression.
Methods
Animals
We purchased C57BL/6J wild-type mice from Guangdong Medical Laboratory Animal Centre. We obtained GAD67-GFP mice from Dr. Shengxi Wu.15 We used male mice aged 9 to 16 weeks for all experiments. Mice were maintained in a pathogen-free, temperature-controlled (22 ± 1°C) mouse facility and housed on a reversed 12-hour light–dark cycle (lights on at 8 am) with 5 to 6 mice per cage; mice had access to food and water ad libitum. All behavioural experiments were performed between 9 am and 6 pm.
Antibodies and viral injections
We obtained anti-NRG1 and anti-RAG antibodies from Genentech and prepared them following a previously published method.8 Briefly, antibodies were dissolved in a saline solution (2 mg/mL) and administered as an intraventricular injection (5 μL) or as a local injection into the dDG (1 μL per side).
Mice were anesthetized with isoflurane (RWD Life Science Co.) using a 5 μL microsyringe (Hamilton) connected to a microsyringe pump (KD Scientific, World Precision Instruments). Then, we injected 400 nL of recombinant adeno-associated viruses (rAAVs; rAAV-CaMKIIα-hM4D[Gi]-mCherry; rAAV-CaMKIIα-hM3D[Gq]-mCherry; rAAV-CaMKIIα-GFP; rAAV-CaMKIIα-hChR2-eYFP) bilaterally into the dDG, at 80 nL/min. To inhibit dDG neurons, a treatment group was injected with rAAV-CaMKIIα-hM4D[Gi]-mCherry and a control group injected with rAAV-CaMKIIα-GFP. To enhance dDG neuronal activity, we used 2 strategies: in the first, a treatment group was injected with rAAV-CaMKIIα-hM3D[Gq]-mCherry and the control group was injected with rAAV-CaMKIIα-GFP; in the second, a treatment group received rAAV-CaMKIIα-hChR2-eYFP injected in the dDG with optofibre implanted in the dDG to stimulate dDG neurons, and a control group was injected with rAAV-CaMKIIα-GFP.
Intraventricular injections were administered to the left lateral ventricle (−0.35 anteroposterior, +1.0 mediolateral, −2.25 dorsoventral) at 1 μL/min for a total volume of 5 μL. Local injections were administered to the dDG (−2.1 anteroposterior, ±1.4 mediolateral, −2.35 dorsoventral) at 200 nL/min for a total volume of 1 μL. All brain region coordinates were relative to Bregma (in mm). After injection, we used a 5 minute rest period to allow virus diffusion.
To ensure adequate viral expression, all behavioural tests were performed at least 4 weeks after viral injections.
Mice that received rAAV-CaMKIIα-hM3D, -hM4D or -GFP injections were given clozapine-N-oxide (CNO, 3 mg/kg i.p.), which potently activates hM3D or hM4D.16 Fear extinction training was performed 30 minutes after CNO injection.
Behavioural tests
Fear conditioning and testing
We conducted fear conditioning and testing following previous methods.8
For fear conditioning, mice underwent 5 trials of a conditioned stimulus (tone; 80 dB, 6000 Hz) co-terminated with a foot shock (0.8 mA, 2 s). We used an intensity of 1 mA in the chemogenetic experiments. Each tone lasted for 30 s, with a 90 s intertrial interval.
For recall tests (1, 2 and 3), mice were placed in a different cage with a different shape (35 × 20 × 20 cm) from the fear conditioning context, and 3 tones were presented with a 90 s intertrial interval. Recall 1 took place 24 hours after fear conditioning to determine the formation of fear memory. To test fear memory expression recalls 2 and 3 took place 24 hours and 10 days after fear extinction, respectively.
For fear extinction training, 15 tones were presented. Each tone lasted for 30 s, with a 60 s intertrial interval. Mice were placed in the same context as for recall tests.
The primary outcome measured was freezing time. Freezing was defined as the complete absence of movement except for normal respiration. Freezing time was defined as the percentage of total time in a state of freezing during tone presentation and was calculated automatically using FreezeFrame software (Coulbourn Instruments).8
Open field test
To test the effect of anti-NRG1, hM3D and deep brain stimulation (DBS) on movement ability, we performed an open field test after recall testing. For the experiments using anti-NRG1 and hM3D, we performed an open field test after recall 2. In the DBS experiment, we performed an open field test after recall 1.
The test was performed in a 50 × 50 × 50 cm chamber, which was cleaned with 75% alcohol after each test. Each mouse started in the centre of the chamber and was allowed to move freely for 15 minutes. Total distance travelled in 15 minutes was recorded and analyzed using ANY-maze software (Global Biotech Inc.).8
Single prolonged stress
To establish a disease model for PTSD, we used a modified procedure for single prolonged stress.17 Mice were restrained for 2 hours in a 50 mL plastic tube with several air holes. After 3 minutes of rest, the mice underwent a 15 minute forced swim test in a glass beaker (15 cm diameter, 30 cm height, 25°C). Mice were dried under a warm lamp, exposed to rat bedding scent for 20 minutes and then exposed to diethyl ether until loss of consciousness. They were then left undisturbed in their home cages with new bedding for 1 week.
All mice underwent fear conditioning training and then were divided into 2 groups with the same freezing levels. The 2 groups received intraventricular injection of anti-NRG1 or anti-RAG and then underwent fear extinction training. We compared freezing levels between the anti-NRG1 and anti-RAG groups during fear extinction training, recall 2 and recall 3 to confirm the effect of anti-NRG1 on fear memory expression in a disease model.
Optogenetic stimulation
To activate the dDG, a treatment group received rAAV-CaMKIIα-hChR2-eYFP injected into the dDG; a control group was injected with rAAV-CaMKIIα-GFP. After injection, both groups of mice were implanted with optic fibre bilaterally in the dDG (200 μm diameter; 0.37 NA; Nanjing Thinker Tech) and neurons in the dDG were stimulated with a 473 nm laser (blue light; 30 s, 8 mW, 10 ms, 20 Hz; from the end of the optic fibers). After fear conditioning, fear extinction training with 15 conditioned stimuli was paired with blue light stimulation. We tested fear memory expression during and after fear extinction training.
Deep brain stimulation
We made twisted bipolar electrodes from insulated silver (0.1 mm diameter) and implanted them bilaterally into the dDG. Stimulating electrodes were implanted bilaterally in the dDG of a control group but never stimulated. After surgery, mice were allowed to recover for 7 days. Then, they underwent fear conditioning with 4 pairings of conditioned stimuli/unconditioned stimuli on day 1. On day 2, mice underwent fear extinction training with 20 conditioned stimuli paired with dDG DBS, consisting of a 300 ms train of square pulses (200 μs pulse width, 100 μA, 100 Hz).18 The DBS was delivered 100 ms after the onset of the conditioned stimulus. We compared freezing levels between the DBS and sham groups in fear extinction and recall tests.
Immunohistochemistry
Mice were killed and brain tissues obtained 90 minutes after the behavioural tests were completed. Sections (30 μm thickness) were cut on a freezing microtome (CM1860 UV; Leica). Immunostaining protocols were taken from previous studies.14 Briefly, sections were treated with 3% H2O2 in phosphate-buffered saline (PBS) for 30 minutes at room temperature, blocked with mouse immunoglobulin G blocking reagent from a Mouse on Mouse Basic Kit (BMK-2202; Vector Laboratories) for 1.5 hours at room temperature and then incubated with mouse anti-c-Fos primary antibody (ab208942; 1:1000; Abcam) overnight at 4°C. Sections were incubated with biotinylated goat anti-mouse secondary antibody (Vectastain ABC Kit; Vector Laboratories) for 1.5 hours at room temperature. Sections were washed with PBS and then incubated with ABC liquid for 2 hours at room temperature. Sections were soaked in DAB colour reagent (SK-4105; Vector Laboratories) until they turned brown, and the reaction was stopped using double-distilled water. Sections were washed in PBS and cover-slipped for imaging.
For double immunostaining of c-Fos and neuronal nuclei (NeuN) or glial fibrillary acidic protein (GFAP), sections were blocked with 10% normal goat serum in PBS with 0.5% Triton-100 at room temperature and incubated with rabbit anti-c-Fos antibody (ab190289; 1:3000; Abcam), mouse anti-NeuN antibody (MAB377; 1:5000; Millipore) or mouse anti-GFAP antibody (MAB360; 1:500; Millipore) overnight at 4°C. Secondary antibodies were Alexa Fluor 488 and 546 conjugated secondary antibodies (goat anti-mouse 488 and goat anti-rabbit 546; 1:400; Invitrogen).
To visualize c-Fos and glutamic acid decarboxylase 67 (GAD67), brain sections from GAD67-GFP mice were blocked with 10% normal goat serum in PBS with 0.5% Triton-100 at room temperature for 1 hour, incubated with rabbit anti-c-Fos antibody overnight at 4°C and then incubated with secondary goat anti-rabbit immunoglobulin G antibody conjugated to Alexa Fluor 546 at room temperature for 1 hour. Sections were then washed in PBS and cover-slipped for imaging.
Data analysis
Statistical analysis was performed using unpaired t tests and 2-way repeated-measures analyses of variance, followed by Bonferroni post-tests (GraphPad Prism software).19 All results are shown as mean ± standard error of the mean, and p < 0.05 was considered statistically significant.
Results
Reduced fear expression and altered fear extinction in mice injected with anti-NRG1 antibodies
We first tested the effect of anti-NRG1 antibodies on fear conditioning and expression of fear memory. Mice underwent fear conditioning 24 hours after intraventricular injection of anti-NRG1 or anti-RAG (control) antibodies (Appendix 1, Figure S1A, available at jpn.ca/200151-a1). Freezing levels in anti-NRG1 mice were significantly lower than in anti-RAG mice on the fourth and fifth trials (Appendix 1, Figure S1B; fourth trial p < 0.001; fifth trial p < 0.05). Recall 10 days after conditioning revealed no difference in freezing levels between the anti-NRG1 and anti-RAG groups (Appendix 1, Figure S1C; recall test 2, t24 = 1.03, p > 0.05), suggesting that fear memory is not altered by anti-NRG1 antibodies: fear expression is altered instead.
Because anti-NRG1 antibodies also engage peripheral targets to induce behavioural alterations that may have interfered with our examination of fear responses,9 we used intraventricular injection to directly introduce antibodies to mouse brains (Figure 1A). To avoid interference, injection was performed after fear conditioning. Freezing levels did not differ between groups injected with anti-RAG and anti-NRG1 antibodies 24 hours after fear conditioning (recall test 1; Figure 1B, t14 = 0.38, p > 0.05). In contrast, at the start of extinction training, freezing levels were significantly lower in the anti-NRG1 group (extinction; Figure 1B, F1,56 = 7.49, p < 0.05; Appendix 1, Figure S2A, F1,28 = 18.03, p < 0.001). This difference in freezing was absent when the same groups were tested 9 days later (recall test 3; Figure 1B, t14 = 1.58, p > 0.05), mostly because of increased freezing levels in the anti-NRG1 group, suggesting that fear memory was unlikely to be altered, but that its expression was suppressed for a period after fear extinction training. To address whether increased locomotion contributed to this effect, we measured total distance travelled in an open field test and found no significant difference between the anti-NRG1 and anti-RAG groups (Figure 1C, t15 = 1.47, p > 0.05).
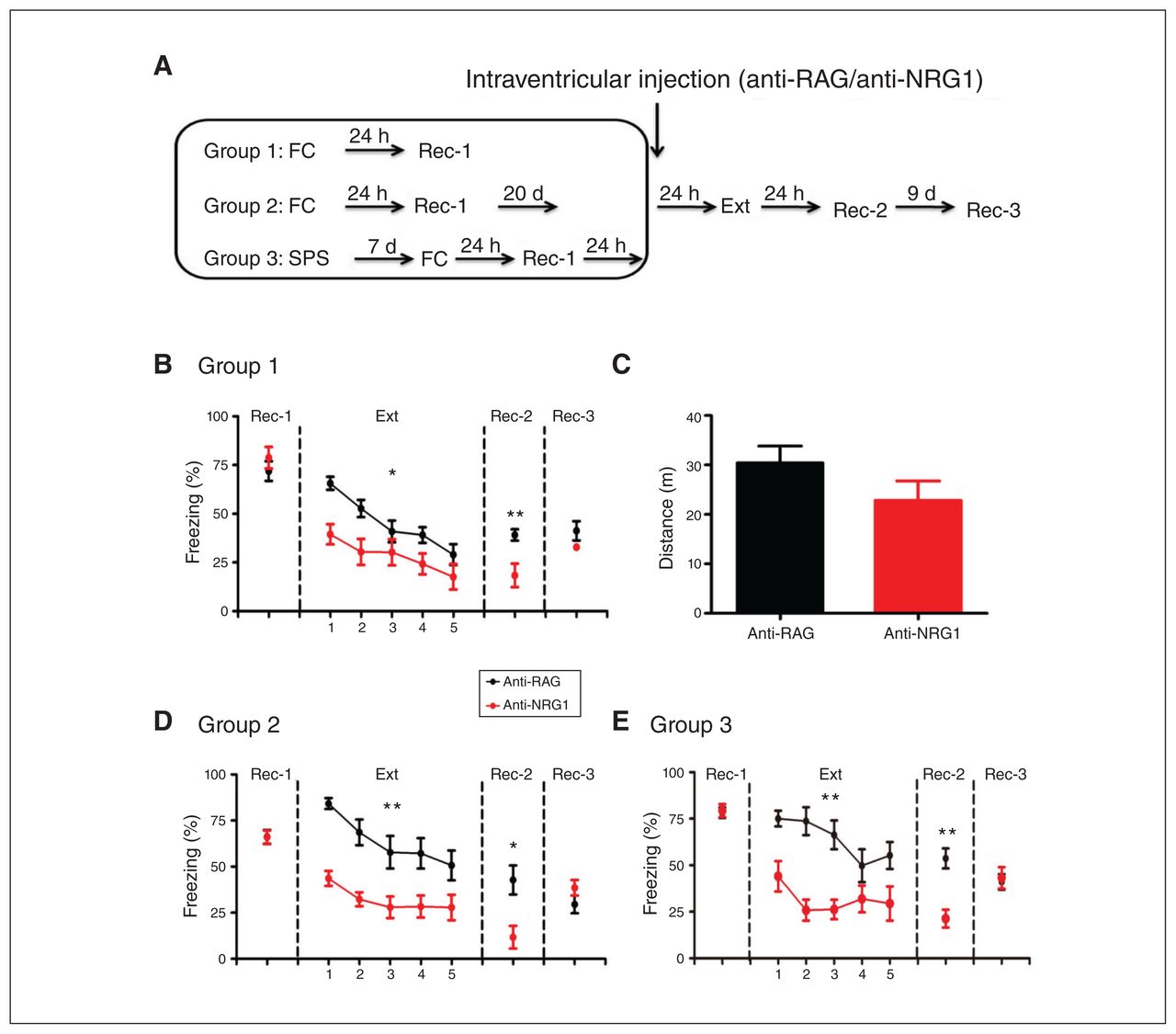
Anti-NRG1 antibody injection led to reduced fear expression. (A) Experimental groups and procedures. (B) Injection of anti-NRG1 antibodies led to lower freezing levels during fear extinction training (anti-NRG1, n = 8 mice; anti-RAG, n = 8 mice). The difference between groups persisted at recall 24 hours after fear extinction training (Rec-2), but not 9 days later (Rec-3). (C) Injection of anti-NRG1 antibodies did not affect locomotion in the open field test (anti-NRG1, n = 8 mice; anti-RAG, n = 9 mice). (D) For the remote fear memory group, injection of anti-NRG1 antibodies led to reduced fear expression (anti-NRG1, n = 6 mice; anti-RAG, n = 6 mice). We observed reduced fear expression at 24 hours after fear extinction training (Rec-2), but not 9 days later (Rec-3). (E) In a single prolonged stress model of post-traumatic stress disorder (group 3), injection of anti-NRG1 antibodies led to reduced fear expression (anti-NRG1, n = 6 mice; anti-RAG, n = 6 mice). We observed reduced fear expression at 24 hours after fear extinction training (Rec-2), but not 9 days later (Rec-3). Freezing score was the average of responses during 3 conditioned stimuli. *p < 0.05; **p < 0.01. Ext = fear extinction training; FC = fear conditioning; Rec-1 = recall 24 hours after fear conditioning; Rec-2 = recall 24 hours after fear extinction training; Rec-3 = recall 9 days after Rec-2; SPS = single prolonged stress.
It is well known that extinction of remote fear memory is much more difficult to achieve.20,21 Thus, we tested whether anti-NRG1 antibodies were more effective in suppressing fear memory after extinction training (Figure 1A, group 2). Similar to our findings for recent fear memory, we found reduced expression of fear memory at both the start of fear extinction training (Appendix 1, Figure S2B, F1,20 = 64.67, p < 0.001) and 24 hours later. Freezing levels were comparable to that of the control group 9 days later (recall test 3; Figure 1D, t10 = 1.41, p > 0.05). We also tested whether anti-NRG1 antibodies could affect expression of fear memory in a well-established model of PTSD (single prolonged stress).17 We found lower freezing levels and lower expression of fear memory in the anti-NRG1 group at the start of extinction training (Appendix 1, Figure S2C, F1,20 = 11. 35, p < 0.01) and 24 hours later (recall test 2; Figure 1E, t10 = 4.47, p < 0.01). However, we observed no difference in freezing levels between the anti-NRG1 and anti-RAG groups 10 days after extinction training (recall test 3; Figure 1E, t10 = 0.30, p > 0.05). Taken together, the above results suggested that the intraventricular injection of anti-NRG1 antibodies were associated with lower expression of fear memory for a period of time after extinction training, but fear memory itself appeared to be unaltered.
Selective elevation in c-Fos expression in the dDG with injection of anti-NRG1 antibodies
To understand which brain regions might contribute to lower expression of fear memory in mice injected with anti-NRG1 antibodies, we examined activated brain regions after fear recall using c-Fos staining (Figure 2A). Mice were killed 24 hours (group 1) or 10 days (group 2) after anti-NRG1/anti-RAG injection. In group 1, we observed a significant increase in the density of c-Fos-positive neurons in the dDG among the few key regions that participate in fear expression and extinction (Figure 2B and C, p < 0.001), and freezing levels were significantly lower in the anti-NRG1 group (Figure 2D, t15 = 7.95, p < 0.001). In group 2, we found no difference between the anti-NRG1 and anti-RAG groups in terms of c-Fos density (Figure 2E, t12 = 0.93, p > 0.05) or freezing levels (Figure 2F, t12 = 1.59, p > 0.05). These results suggested that the density of activated dDG cells may be related to freezing levels. We also found that the density of activated dDG cells was significantly higher in mice injected with anti-NRG1 than in those injected with anti-RAG in a novel experiment, but no difference between the 2 groups in the home cage (Appendix 1, Figure S3). To identify the cell types activated, we used c-Fos staining in GAD 67-GFP mice (Figure 2G) or double staining of c-Fos with GFAP (a marker of astrocytes; Figure 2H), but we found virtually no colocalization with these markers, indicating that c-Fos neurons were not GABAergic neurons or astrocytes. We also stained brain sections with anti-NeuN and anti-c-Fos simultaneously (Figure 2I) and confirmed that c-Fos-positive cells were neurons (Figure 2J). The above results indicated that excitatory neurons in the dDG were the main target for activation in mice injected with anti-NRG1 antibodies.

Intraventricular injection of anti-NRG1 antibodies led to reduced fear expression and activated excitatory neurons in the dentate gyrus. (A) Experimental procedures. (B) Quantification of c-Fos-positive cells in a few key brain regions. We found a significant difference in the dDG (anti-NRG1, n = 9 mice; anti-RAG, n = 8 mice). (C) Sample images of c-Fos immunostaining in mice injected with anti-RAG (left) or anti-NRG1 (right) antibodies. Scale bar, 100 μm. (D) Injection of anti-NRG1 antibodies suppressed fear expression at 24 hours after injection (anti-NRG1, n = 9 mice; anti-RAG, n = 8 mice). (E) Injection of anti-NRG1 antibodies had no effect on the density of c-Fos-positive neurons 10 days after injection (anti-NRG1, n = 7 mice; anti-RAG, n = 7 mice). (F) Injection of anti-NRG1 antibodies had no effect on fear expression 10 days after injection (anti-NRG1, n = 7 mice; anti-RAG, n = 7 mice). (G) Sample images for c-Fos and GAD67 staining. Scale 100 μm. (H) Sample images for c-Fos and GFAP staining. Scale 100 μm. (I) Sample images for c-Fos and NeuN staining. Scale 100 μm. (J) Colocalization between c-Fos and NeuN, GAD67 or GFAP. ***p < 0.001. BLA = basolateral amygdala; dCA1 = dorsal hippocampus subfield 1; dCA3 = dorsal hippocampus subfield 3; dDG = dorsal dentate gyrus; FC = fear conditioning; GAD67 = glutamic acid decarboxylase 67; GFAP = glial fibrillary acidic protein; mPFC = medial prefrontal cortex; NeuN = neuronal nuclei; NS = not significant; PVT = paraventricular thalamus; Rec-1 = recall 24 hours after fear conditioning; Rec-2 = recall 10 days after fear extinction training.
Reduced fear expression with elevated neural activity in the dDG
If the dDG is a key brain region regulating expression of fear memory, we would expect that local injection of anti-NRG1 antibodies into the dDG would have a similar effect. We injected anti-NRG1 antibodies into the dDG bilaterally after fear conditioning and the first recall test (Figure 3A; 1 μL/side). We first quantified c-Fos-positive neurons in the dDG (Figure 3B) and confirmed a significantly higher density of c-Fos-positive neurons in the dDG of mice injected with anti-NRG1 antibodies (Figure 3C, t20 = 6.38, p < 0.001). Expression of fear memory was lower compared to the anti-RAG group (extinction; Figure 3D, F1,80 = 12.06, p < 0.01). Expression of fear memory was also significantly lower at 24 hours (recall test 2; Figure 3D, t20 = 2.82, p < 0.05) and 9 days later (recall test 3; Figure 3D, t20 = 2.21, p < 0.05) after extinction training in the anti-NRG1 group. Closer examination revealed that the difference between these 2 groups was due in particular to increased freezing levels in the anti-RAG group, which was absent in the other groups (e.g., Figure 1B, D and E). The effect on fear expression of direct injection of anti-NRG1 antibodies into the dDG may have been because of the density of activated neurons in the dDG. Hence, local injection of anti-NRG1 antibodies into the dDG mimicked intraventricular antibody injection in terms of expression of fear memory and dDG activation.

Injection of anti-NRG1 antibodies led to reduced fear expression and elevated c-Fos expression in the dentate gyrus. (A) Experimental procedures. (B) Sample immunostaining images of c-Fos in mice injected with anti-RAG (left) or anti-NRG1 (right) antibodies. (C) Quantification of the density of c-Fos-positive neurons in the dDG in mice injected with anti-NRG1 or anti-RAG antibodies (anti-NRG1, n = 12 mice; anti-RAG, n = 10 mice). (D) Injection of anti-NRG1 antibodies in the dDG led to lower fear expression without affecting initial freezing levels. Freezing levels were significantly lower in the anti-NRG1 group 24 hours after extinction training (Rec-2) and 9 days later (Rec-3; anti-NRG1, n = 11 mice; anti-RAG, n = 11 mice). Freezing score was the average of responses during 3 conditioned stimuli. *p < 0.05; **p < 0.01; ***p < 0.001. dDG = dorsal dentate gyrus; Ext = fear extinction training; FC = fear conditioning; Rec-1 = recall 24 hours after fear conditioning; Rec-2 = recall 24 hours after fear extinction training; Rec-3 = recall 9 days after Rec-2.
Anti-NRG1 antibody effect blocked by DREADD inhibition and mimicked by DREADD excitation of dDG neurons
If elevated activity in the dDG is required for lower expression of fear memory in mice injected with anti-NRG1 antibodies, then inhibiting neural activity in the dDG should eliminate the antibody effect. To test this, we injected an inhibitory hM4D receptor-mCherry fusion virus (rAAV-CaMKIIα-hM4Dm-Cherry) into the dDG of anti-NRG1 and anti-RAG mice, and GFP reporter virus (rAAV-CaMKIIα-GFP) into the dDG of a control group (Figure 4A). Twenty-four hours after injection of the anti-NRG1 antibodies, CNO was administered (i.p., 3 mg/kg) to inhibit dDG neurons, followed by extinction training in 30 minutes. We found no difference in freezing levels or their rate of reduction in the anti-NRG1 and anti-RAG groups expressing the hM4D virus (Figure 4B; extinction F1,80 = 3.08, p > 0.05; recall test 2 t20 = 0.43, p > 0.05). In mice injected with the GFP virus, the anti-NRG1 group showed lower freezing levels than the anti-RAG group (Figure 4C; extinction F1, 72 = 18.51, p < 0.001; recall test 2 t18 = 2.17, p < 0.05). These results demonstrated that hM4D-mediated inhibition of dDG excitatory cells abolished the effect of anti-NRG1 antibody injection on the expression of fear memory.
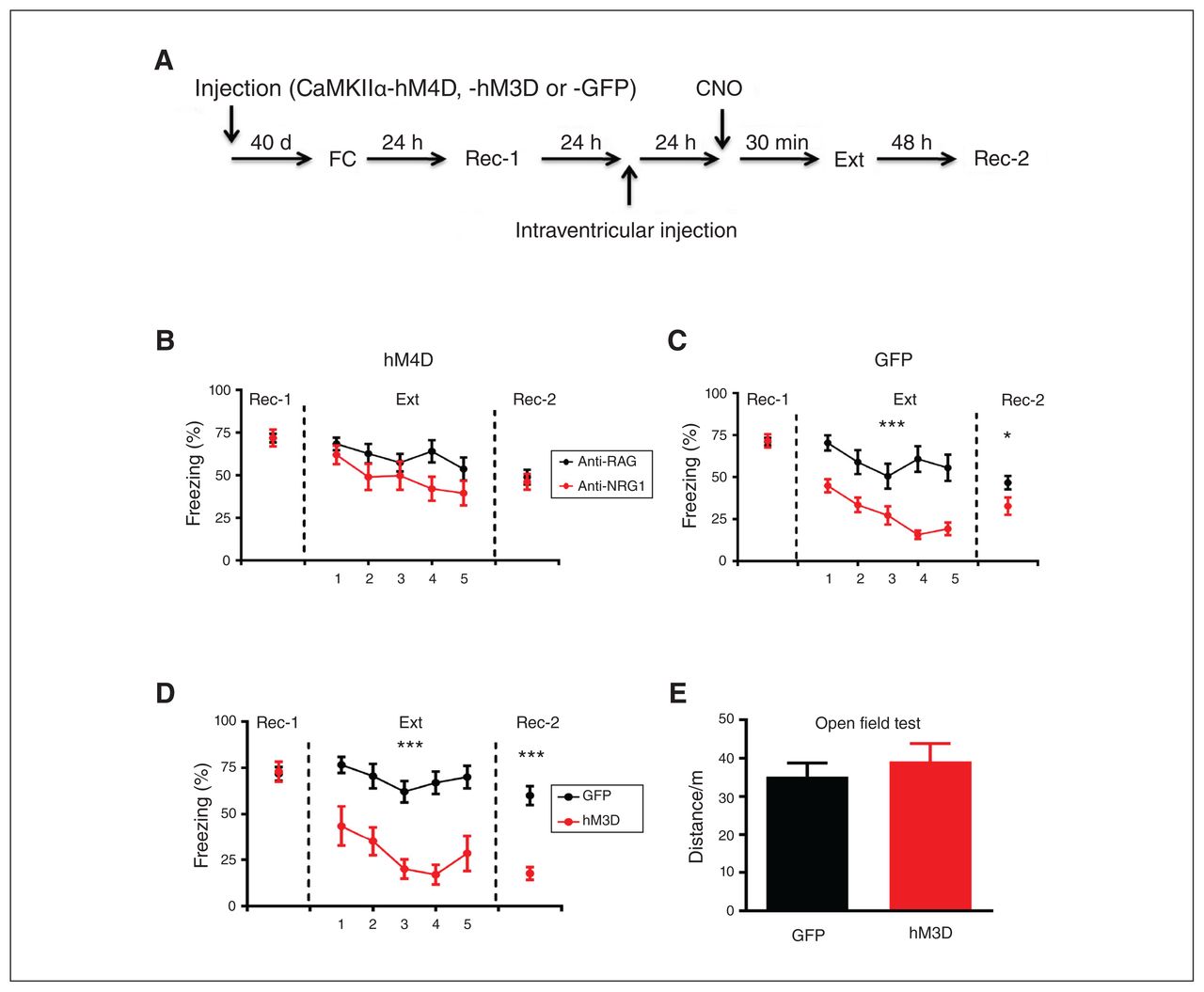
DREADD modulation of neuronal activity in the dDG affected fear expression. (A) Experimental procedures. (B) Inhibition of dDG activity with an inhibitory CaMKIIα-hM4D-mCherry fusion protein abolished the reduction in fear expression induced by anti-NRG1. Freezing levels were not significantly different between the 2 groups at 24 hours after fear extinction training (anti-NRG1, n = 10 mice; anti-RAG, n = 12 mice). (C) Expression of CaMKIIα-GFP in the dDG had no effect on the reduction of fear expression induced by anti-NRG1. Freezing levels were significantly lower in the anti-NRG1 group than in the anti-RAG group (anti-NRG1, n = 9 mice; anti-RAG, n = 11 mice). (D) Excitation of the dDG led to reduced fear expression. We also observed reduced fear expression 48 hours after fear extinction training (hM3D, n = 8 mice; GFP, n = 11 mice); ***p < 0.001. (E) Increased neuronal activity in the dDG with an excitatory CaMKIIα-hM3D-mCherry fusion protein did not affect locomotion in the open field test (hM3D, n = 8 mice; GFP, n = 8 mice). Freezing score was the average of responses during 3 conditioned stimuli. *p < 0.05; ***p < 0.001. CaMKIIα-GFP = calcium/calmodulin-dependent protein kinase II α green fluorescent protein; CaMKIIα-hM3D = calcium/calmodulin-dependent protein kinase II α human muscarinic-3 receptor DREADD-mutation; CaMKIIα-hM4D = calcium/calmodulin-dependent protein kinase II α human muscarinic-4 receptor DREADD-mutation; dDG = dorsal dentate gyrus; DREADD = designer receptor exclusively activated by designer drugs; Ext = fear extinction training; FC = fear conditioning; Rec-1 = recall 24 hours after fear conditioning; Rec-2 = recall 48 hours after fear extinction training.
Next, we conducted a contrasting experiment by expressing excitatory hM3D receptor-mCherry fusion protein (rAAV-CaMKIIα-hM3D-mCherry) into the dDG of wild-type mice, and a GFP reporter virus (rAAV-CaMKIIα-GFP) into the dDG of control mice (Figure 4A). The rationale was that enhancing dDG neuronal activity using hM3D should mimic the effect of anti-NRG1 injection in wild-type mice. Compared to the GFP group, we found significantly lower freezing levels in the h3MD group (Figure 4D; extinction F1,68 = 30.19, p < 0.001; recall test 2 t17 = 6.37, p < 0.001), resembling the effects in mice injected with anti-NRG1 antibodies. We found that CNO injection did not alter locomotion in an open field test, and we found no difference between the hM3D and GFP groups in terms of total distance travelled (Figure 4E, t14 = 0.66, p > 0.05). We found c-Fos-expressing cells in the dDG after CNO injection in the hM3D group, but not in the hM4D group (Appendix 1, Figure S4). Thus, hM3D-mediated activation of dDG excitatory cells suppressed expression of fear memory.
Suppressed expression of fear memory with stimulation of dDG neurons
To further confirm the effect of activation of the dDG on fear expression and fear extinction, mice injected with adeno-associated viruses (rAAV-CaMKIIα-hChR2-eYFP or rAAV-CaMKIIα-GFP) underwent auditory fear conditioning (Figure 5A). After fear conditioning, the 2 groups of mice had similar freezing levels during recall test 1. However, when the mice were exposed to the conditioned stimulus with blue light delivered to the bilateral dDG using optical fibres, freezing levels in the hChR2 group were significantly lower than levels in the GFP group. As well, expression of fear memory was significantly lower at 24 hours and 10 days after extinction training in the hChR2 group (Figure 5B; extinction F1,90 = 57.85, p < 0.001; recall test 2 t15 = 3.05, p < 0.01; recall test 3 t15 = 5.57, p < 0.001), resembling the effect of hM3D-mediated activation of dDG excitatory cells. We also tested the effect of optostimulation in dDG and found numerous c-Fos-expressing cells in the dDG after blue light stimulation (Appendix 1, Fig. S4).

Stimulation of dDG neurons resulted in reduced fear expression. (A) Experimental procedures. (B) Optogenetic activation of dDG neurons led to reduced fear expression. We also observed reduced fear expression 24 hours after extinction training (hChR2, n = 8 mice; GFP, n = 9 mice). (C) Experimental procedures. (D) Fear expression was suppressed in the DBS group during both fear extinction training and recall test 1 (n = 9 mice; sham, n = 6 mice). (E) DBS in the dDG did not affect locomotion measured in the open field test (unpaired t test, t8 = 0.35, p > 0.05; DBS, n = 9 mice; sham, n = 8 mice). *p < 0.05; **p < 0.01; ***p < 0.001. Freezing score was the average of responses during 2 conditioned stimuli in B and during 3 conditioned stimuli in D. CaMKIIα-GFP = CaMKIIα-GFP = calcium/calmodulin-dependent protein kinase II α green fluorescent protein; CaMKIIα-hChR2 = calcium/calmodulin-dependent protein kinase II α H134R mutation in channelrhodopsin-2; DBS = deep brain stimulation; dDG = dorsal dentate gyrus; Ext = fear extinction training; FC = fear conditioning; Rec-1 = recall 24 hours after fear conditioning; Rec-2 = recall 48 hours after fear extinction training; Rec-3 = recall 9 days after Rec-2.
We also conducted a DBS experiment to modulate the activity of the dDG (Figure 5C). Two weeks after surgery, mice were fear-conditioned and tested. Freezing levels were significantly lower during both extinction and recall in the DBS group than in the sham group (Figure 5D; F1,52 = 10.46, p < 0.01; recall test 1 t13 = 2.39, p < 0.05), but locomotion in the open field test was not different between the 2 groups (Figure 5E; t15 = 0.56, p > 0.05). After DBS in the dDG, we also found abundant c-Fos-expressing cells in the dDG (Appendix 1, Figure S4). Taken together, the above results showed an effective reduction in fear expression using DBS in the dDG.
Discussion
In this study we examined the contribution of the dDG to fear expression. We found the following: intraventricular injection of anti-NRG1 antibodies before fear extinction was associated with lower freezing levels in fear-conditioned naive mice, PTSD model mice, and testing of remote fear memory; intraventricular injection of anti-NRG1 antibodies selectively activated excitatory neurons in the dDG; local injection of anti-NRG1 antibodies in the dDG activated dDG neurons and reduced fear expression; hM4D-mediated inhibition of the dDG blocked anti-NRG1 effects on fear expression; hM3D or optogenetic activation of the dDG suppressed fear expression; and DBS in the dDG reduced fear expression. These findings support the idea that elevated activity in the dDG suppress fear expression.
The major finding of the current study is that activation of the dDG can reduce the expression of auditory fear memory. This finding was consistent with those of our previous work: that Dnmt3a overexpression in the dDG prevents fear renewal and elevates the activity of dDG, and that knockdown of Dnmt3a in the dDG promotes fear renewal.14 Overexpression of Dnmt3a in the dDG resulted in higher density of c-Fos neurons in the dDG.14 This result means that the activity of the dDG is associated with expression levels of Dnmt3a in the dDG. Mice with Dnmt3a overexpression in the dDG showed no significant fear renewal, supporting our finding in the present study about a correlation between fear expression and the activity of the dDG.
The exact contribution of the dorsal hippocampus to auditory fear conditioning is poorly understood, especially with respect to the contribution of the dDG. Although some studies have suggested that modulating the activity of the dorsal hippocampus did not affect auditory fear conditioning,11,22,23 Quinn and colleagues24 and Bast and colleagues25 both found that freezing levels during tone presentation were significantly reduced after neurotoxic lesion of the dorsal hippocampus. Activating dDG neurons that were tagged during auditory fear conditioning was sufficient to trigger expression of that memory.26 Before fear extinction training, inactivation of the dorsal hippocampus reduced the rate of extinction. 27 Thus, the dorsal hippocampus is implicated in auditory fear conditioning and extinction. Our finding that dDG activation suppressed fear expression during memory recall provides the most direct evidence for the contribution of the dDG to cued expression of fear memory.
The dDG granule cells receive their major inputs from the entorhinal cortex (EC) and then project to the hippocampal subfield 3 (CA3). In turn, the CA3 pyramidal cells project to the hippocampal subfield 1 (CA1), which projects back to the EC, creating a synaptic loop (EC–DG–CA3–CA1–EC). The CA3 region also projects to the lateral septum,28 which is activated after auditory fear conditioning,29 and pretraining inactivation of the lateral septum abolishes learned fear response.30 The lateral septum has strong bidirectional interaction with the amygdala and hypothalamus, and both structures play an important role in auditory fear learning.29,31,32 Taking these findings together, it is possible that the dDG suppresses fear response by directly regulating the expression of fear memory, via CA3 projection to the lateral septum and subsequently to the amygdala. Electrical stimulation of the lateral septum had a predominantly inhibitory effect on neurons in the amygdala, 33 but whether this effect is mediated or can be mimicked by CA3 inputs to the lateral septum is unknown.
The dDG also receives inputs from the locus coeruleus and diffuse projections from the ventral tegmental area, which participate in regulating novelty detection.34–36 It has been demonstrated that the dorsal hippocampus also participates in novelty-related exploration.37–40 Moncada and colleagues39 found that previous exposure to a novel environment promoted long-term memory formation. The dDG also plays a role in novelty detection,41 and activating parvalbumin neurons in the dDG appears to impair social interaction induced by novelty.23 Some studies have shown that novelty exposure before fear extinction training facilitated and strengthened fear extinction.42–45 Rats exposed to a novel environment for 5 minutes before fear extinction training showed enhanced fear extinction, which was fully blocked by anisomycin in the dorsal CA1.46 Whether elevated activation of the dDG by novelty exploration can modulate the expression of fear memory requires further investigation.
Fear memory did not appear to be altered by anti-NRG1 antibody injection. The effect of dDG activation may thus be limited to modulating the expression of conditioned fear. If this is the case, one advantage of targeting the underlying mechanism or system is to suppress or modulate fear expression while preserving the fear memory. To explore the possibility of manipulating the dDG dynamically with methods transferable to human treatment, we tested DBS. We found that freezing levels in mice that received DBS in the dDG during fear extinction were significantly lower than in mice from the sham group; DBS may help to suppress fear expression during fear extinction training. Although DBS has been used routinely to treat depression47 and obsessive–compulsive disorder48 and preclinical DBS studies in fear conditioning animal models have suggested its potential use for treatment-resistant PTSD,49–51 very few studies have explored the clinical effect of DBS in PTSD.52–54 About 20% to 30% of PTSD patients do not respond to medications and conventional psychotherapy.49,51 A recent clinical trial showed that 6 patients with treatment-resistant PTSD experienced a 30% decrease in clinician-administered PTSD scale scores after bilateral basolateral amygdala high-frequency stimulation.52,54 Our results supported DBS as a potential therapeutic tool for refractory PTSD.
Limitations
We found that elevating dDG activity suppressed the expression of fear memory, but we did not investigate the neural circuits underlying this phenomenon. We propose that this suppression occurs via CA3 projection to the lateral septum and then the amygdala. We also did not address whether fear memory was still intact with dDG activation.
Conclusion
We have provided evidence that the dDG may modulate auditory fear conditioning, in that dDG activation suppressed the expression of conditioned fear. Because reduced fear expression is seen after DBS in the dDG, we suggest that modulating dDG activity may have therapeutic potential in treating diseases that involve aberrant emotional responses, such as PTSD.
Acknowledgements
We thank T. Li for the virus injection and Dr. Z. Gong for suggestions on immunohistochemistry methods. This work is supported by grants (2019SHIBS0004, SZBL2019062801003, KQTD2015032709315529, SZSM201812005).
Footnotes
Competing interests: None declared.
Contributors: Y. Zhang and Q. Zhou designed the study. Y. Zhang, Z. Wang, J. Ju and J. Liao acquired the data, which Y. Zhang, Z. Wang, J. Ju and Q. Zhou analyzed. Y. Zhang and Q. Zhou wrote the article, which all authors reviewed. All authors approved the final version to be published and can certify that no other individuals not listed as authors have made substantial contributions to the paper.
- Received August 7, 2020.
- Revision received November 2, 2020.
- Revision received December 18, 2020.
- Accepted January 23, 2021.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is noncommercial (i.e., research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools


