

Abstract
The pathophysiology of schizophrenia and bipolar disorder involves a complex interaction between genetic and environmental factors that begins in the early stages of neurodevelopment. Recent advancements in the field of induced pluripotent stem cells (iPSCs) offer a promising tool for understanding the neurobiological alterations involved in these disorders and, potentially, for developing new treatment options. In this review, we summarize the results of iPSC-based research on schizophrenia and bipolar disorder, showing disturbances in neurodevelopmental processes, imbalance in glutamatergic–GABAergic transmission and neuromorphological alterations. The limitations of the reviewed literature are also highlighted, particularly the methodological heterogeneity of the studies, the limited number of studies developing iPSC models of both diseases simultaneously, and the lack of in-depth clinical characterization of the included samples. Further studies are needed to advance knowledge on the common and disease-specific pathophysiological features of schizophrenia and bipolar disorder and to promote the development of new treatment options.
Introduction
Schizophrenia and bipolar disorder are complex and challenging mental disorders, whose etiopathogenesis can be traced back to a complex interaction between genetic and environmental factors that begins at the early stages of neurodevelopment.1–4
Together, these disorders affect about 2%–3% of the population worldwide5–7 and are associated with severe functional impairment, a remarkable reduction of quality of life, high levels of disability, and premature death.8–10
Schizophrenia is a psychotic disorder whose onset is typically in adolescence or early adulthood between the ages of 16 and 30 years.11 The disorder is characterized by a broad and diverse spectrum of clinical manifestations, including formal thought disorder, delusions, hallucinations, negative symptoms, and deficits in cognitive and social functioning.12–15 Importantly, although the reduction of symptom severity contributes to functional recovery, patients with remission of psychotic symptoms may still present serious impairment in different areas of real-life functioning, mainly because there are no effective treatments for major determinants of poor outcomes such as negative symptoms and deficits in cognition.16–18
Bipolar disorder is characterized by alternating and fluctuating episodes of depression and mania or hypomania, or mixtures of manic and depressive features.19,20 The onset of bipolar disorder is between the ages of 20 and 30 years, with patients displaying heterogeneous clinical presentations and response to treatments.20–23
Despite distinct diagnostic criteria, schizophrenia and bipolar disorder show some degree of overlap on different aspects, especially in certain subpopulations of patients.24–29 The first of these aspects pertains to the clinical presentation of these 2 disorders. Specifically, within the spectrum of psychotic disorders, the diagnosis of schizoaffective disorder includes clinical features of schizophrenia and bipolar disorder, and can be subdivided into bipolar and depressive types, the former characterized by the presence of manic episodes.30 Furthermore, bipolar disorder is commonly divided into 2 subtypes with distinct features, type I and type II. Both types can present psychotic symptoms, but these are generally more frequent among patients with type I bipolar disorder.31 Finally, cognitive disturbances, can be frequently recorded in schizophrenia but are also present in more subtle forms in both types of bipolar disorder, with some studies reporting a higher frequency of these deficits in type I bipolar disorder.27,32–35
Alterations at the embryonic and later neurodevelopmental stages — leading to defects in neuronal differentiation, connectivity, and neurotransmission — have been traced both in schizophrenia and bipolar disorder, with higher frequencies recorded in schizophrenia.2,36–38 Nevertheless, aberrant neurodevelopmental trajectories have been consistently recorded in a subset of people with bipolar disorder, particularly those with an early age of illness onset and those exhibiting psychotic symptoms.2
Schizophrenia and bipolar disorder seem to share some identified genetic and environmental risk factors that seem to underpin the neurobiology of both disorders.39–42 For instance, a study involving 2 million families showed a remarkable increase in risk of developing schizophrenia if a close relative was diagnosed with bipolar disorder.40 Specifically, several common single nucleotide polymorphisms (SNPs) across the genome have been detected for schizophrenia and bipolar disorder, 43 with remarkably high estimates of genetic correlation between these 2 disorders compared with other psychiatric conditions.43,44 Interestingly, patients with type I bipolar disorder and patients with bipolar disorder with psychotic features present similar genetic risk factors as patients with schizophrenia compared with patients with type II bipolar disorder, who are more genetically correlated with patients with major depressive disorder.43,45–47 Furthermore, rare genetic variants, such as copy number variations, have been linked more to the manifestation of schizophrenia than bipolar disorder, with loci overlapping with autism spectrum disorders and intellectual disability.48 Loci of copy number variations have only rarely and inconsistently been associated with bipolar disorder.49 However, copy number variations have been implicated in the pathogenesis of bipolar disorder in specific cases such as among patients with schizoaffective disorder bipolar type.50 Overall the different SNPs or copy number variations associated with schizophrenia and bipolar disorder have been found to affect the expression of genes regulating glutamate neurotransmission,43,51–54 sodium and calcium signalling,55–57 cytoskeletal components,58,59 or cortical neurogenesis.60 Furthermore, genetic background seems to interact with common environmental factors61–63 that may increase the risk for both disorders,1,3 such as perinatal risk factors, 64,65 adverse life events (especially during childhood),66,67 and the misuse of certain substances or drugs.
Structural and functional neuroimaging studies have also identified similarities in the brain correlates of schizophrenia and bipolar disorder, such as smaller total brain and hippocampal volumes, thinner cortical thickness, and larger ventricular volumes.68–75 However, effect sizes of these alterations were smaller in bipolar disorder than schizophrenia; these disorders also show some disease-specific brain abnormalities such as distinct grey matter reductions in the pregenual cingulate cortex (anterior Brodmann area 4) in bipolar disorder and larger intracranial volume and thicker right parietal cortex in schizophrenia.69,76–79
Despite the important advancements deriving from studies involving animal models, genetic sequencing, neuroimaging, and postmortem observations, deciphering the pathophysiology of these disorders represents still a challenge. 26,80–84 Recent progress in the field of stem cell research offers a promising avenue for understanding the neurodevelopmental and molecular alterations involved in these disorders and, potentially, for developing new treatment options. 85 The advent of induced pluripotent stem cells (iPSCs) in 2006 has allowed the creation of in vitro models that retain the genetic information of the original donor and can help detect the presence of any aberrations in the molecular cascades present in a wide array of psychiatric disorders.86 These cells are obtained using a set of transcription factors to induce adult somatic cells to return to an embryonic-like state, regaining pluripotency and the ability to differentiate into various cell types such as neural progenitor cells (NPCs), fully developed neurons, microglia, and astrocytes.86–90 Three-dimensional (3D) models, organoids, have also been developed, whereby iPSCs growing in matrix substrates are able to recapitulate, quite successfully, the functional and structural development of the adult nervous system.91–93
Overall, studies employing iPSCs in schizophrenia and bipolar disorder have adopted 2 main approaches. The first focuses on the genetic risk variants associated with these disorders and how the presence of these allelic variations in iPSC models affect molecular and cellular characteristics. The second focuses on the differences in neurodevelopmental processes, morphological characteristics, and neurotransmission recorded in iPSC-derived cells of patients and healthy donors.
Therefore, the aim of the current review was to provide a comprehensive overview on results obtained with both approaches, highlighting how iPSC studies have advanced understanding of main pathological alterations linked to schizophrenia and bipolar disorder, and the shared or specific mechanisms traced in the 2 disorders. For the current narrative review, we searched PubMed for articles in English published until March 2023 for combinations of schizophrenia, psychotic disorders, or bipolar disorder and stem cells or iPSC. We employed 10 narrative reviews and studies from their bibliographies in the current review to provide a detailed description of the literature addressing this topic.21,91,94–101 Furthermore, we retrieved and included additional studies and recent publications from the PubMed search. Findings from these studies have been arranged under different sections that describe how the iPSC models have provided further evidence on the pathophysiological mechanisms linked to schizophrenia and bipolar disorder. We discuss the shared and distinct alterations detected in the 2 disorders and the current challenges for iPSC studies.
Alterations in neurodevelopmental trajectories
The neurodevelopmental hypothesis postulates that disturbances in neurogenesis, occurring both during embryonic development and the postnatal period, can functionally alter neural circuitry and lead to the onset of schizophrenia and bipolar disorder.36,102 Neurogenesis involves cell proliferation, differentiation, programmed cell death, and segregation into specific regions, which all contribute to the development of the components of the nervous system.103 Molecular biology, neuroimaging, and postmortem studies have supported the hypothesis of disturbances in neurogenesis, showing that several genes associated with schizophrenia and bipolar disorder are expressed prenatally and are involved in cell differentiation, leading to alterations in cortical structures during early neurodevelopmental stages or at the onset of the disease. 2,104,105 For instance, some of the genetic risk factors of these 2 disorders are mostly expressed mid-pregnancy,106 a period when the occurrence of adverse events in the birthing parent’s life may cause aberrant development of key brain structures such as the hippocampus, limbic system, and prefrontal cortex (PFC), leading to mental health disorders in adult offspring.107,108
One of the molecular pathways that has gained attention in studies using stem cells to explore the neurodevelopmental hypothesis is the Wnt signalling pathway.60,93,109–113 The Wnt pathway is involved in cellular communication, proliferation, migration, and polarity, as well as in embryonic and adult stem cell homeostasis and fate determination throughout embryogenesis and organogenesis.114–116 Specifically, during embryogenesis, the Wnt signalling pathway plays an important role in neurogenesis, inhibiting the differentiation of NPCs into oligodendrocytes progenitor cells.115
Schizophrenia models
Different alterations of the Wnt pathway in iPSC from patients with schizophrenia have been reported. Studies have found an association between sequence variations in the DISC1 (disrupted in schizophrenia) gene and alterations in Wnt signalling. The DISC1 gene is one of the most studied in schizophrenia research given the frequent associations reported between its allelic variations and the development of schizophrenia.117–119 Moreover, some studies have also reported an association between genetic variations of DISC1 and bipolar disorder.120,121
As a multifunctional scaffold protein, DISC1 interacts with several proteins involved in neuronal migration, neurite growth, proliferation of NPCs, and synaptic function.117,122 The effects of DISC1 mutations on the Wnt pathway and how these could be linked to defects in neurodevelopmental trajectories can be investigated with iPSC models. For instance, a study investigated abnormalities in Wnt signalling using NPCs engineered with a DISC1 exon 2/8 interruption gene in iPSCs from healthy donors.111 The study showed that these NPCs had an increase in Wnt signalling activity, with alterations in the expression of neuronal fate–related genes (i.e., increased expression of dorsal progenitor markers and decreased expression of ventral progenitor markers), possibly driving aberrations in the distribution of cell types generated during cortical development.111 In line with these results, another study comparing 3D organoid models presenting either a mutation of DISC1 in exon 8 or wild-type DISC1, showed a decrease in NPC proliferation and alterations of ventricular structures, underpinned by an increase in baseline Wnt signalling, in the DISC1-mutated model.93 In contrast to these results, a decrease in Wnt signalling activity was recently detected in iPSC-derived brain organoids from patients with schizoaffective disorder and schizophrenia.109 Specifically, this decrease in Wnt signalling led to an increase in differentiation into neurons that produce γ-aminobutyric acid (GABA), with elevated inhibitory synaptic density, a reduced proliferation of NPCs, and faster neuronal maturation. 109 Therefore, discrepancies of findings concerning Wnt levels might be owing to 2 main factors. The first factor concerns the limitations in reproducing the dynamics of the developing brain in organoids models since, in the brain, excitatory and inhibitory neurons are generated in separate brain regions, which can be only partially reproduced in 3D iPSC models.94 The second factor is related to the fact that the 2 organoid models were derived from 2 different research protocols. In fact, one study focused on the cellular phenotypes in twins discordant for schizophrenia diagnosis to detect disease-specific alterations, while minimizing inter-individual genetic variations.109 On the other hand, the study by Srikanth and colleagues93 investigated the consequences of DISC1 interruption in cells derived from healthy donors to determine which subtle neurodevelopmental disruption might be linked to DISC1 and predispose to psychiatric disorders.
A study comparing the characteristics of NPCs derived either from iPSCs with DISC1 mutation or iPSCs with wild-type DISC1 showed that alterations in neurogenesis in the former might also be owing to defects in microRNA regulation, such as an increase in microRNA-219 levels.123 MicroRNAs are a class of small, noncoding RNAs that act as post-transcriptional regulators of gene expression. Specifically, microRNA-219 inhibits NPC proliferation and self-renewal by controlling the expression of genes involved in these processes. 123 Therefore, alterations in DISC1 expression and increases in microRNA-219 could be linked to a reduction in neural stem cell proliferation. Recent studies have also revealed that mutations in DISC1 may lead to aberrant interactions with other binding proteins.122,124 One study highlighted the importance of the interaction between DISC1 and Ndel1, which is a component of the dynein complex, implicated in neurogenesis.125 This process was studied in human brain organoid models of patients with schizophrenia with DISC1 mutations, showing a reduction in neural stem cell proliferation related to disruption of DISC1–Ndel1 interaction and delayed cell-cycle progression in radial glial cells.126 Overall, these studies seem to suggest that disruptions in Wnt signalling pathway due to DISC1 mutations might cause a reduction in proliferation of neuronal cells, leading to the development of schizophrenia. This hypothesis is in line with postmortem and brain imaging evidence that shows reduction of brain volume among patients affected by schizophrenia.127,128
Another genetic risk factor that has been strongly linked to schizophrenia and neurodevelopmental abnormalities is the 22q11.2 deletion,129 with a prevalence of this mutation in 0.5%–1% of patients with schizophrenia.130,131 This genetic mutation has also been associated with mood disorders ( major depressive disorder and bipolar disorder), cognitive impairments, and developmental delays.132–134 Furthermore, the 22q11.2 deletion has been associated with microRNA-mediated pathways.135–137 For instance, iPSC studies focusing on the 22q11.2 deletion showed remarkable alterations in DCGR8 gene expression, which encodes a protein that mediates the biogenesis of microRNAs, resulting in alterations of neural differentiation efficiency, neurite outgrowth, and cellular migration.135–137 A further insight on the alterations linked to this mutation was provided by iPSCs generated from 1 patient with schizophrenia with 22q11.2 deletion.138 In this study, the obtained neurospheres — free-floating clusters of neural stem cells generated from iPSCs — showed reductions in their size upon differentiation in the culture derived from the patient with this genetic deletion, without alteration in their total number.138 In addition, an enrichment of apoptotic genes was observed in iPSCs from the patient with schizophrenia, which might be linked to increased susceptibility to apoptosis in early neurodevelopmental processes and lead to synaptic or dendritic loss.
Overall, the current results of iPSC models of schizophrenia have shown how alterations traced in levels of Wnt, molecular signalling cascades, and the concentration of microRNAs might lead to defects in different neurodevelopmental processes such as neuronal proliferation, maturation, and migration.
Bipolar disorder models
Similar evidence of altered Wnt pathways has been found in iPSC-derived neurons of patients with bipolar disorder.139–141 In particular, a study reported an altered expression of several genes related to the Wnt pathway and neurodevelopment such as PAX6 in iPSCs of patients affected by bipolar disorder, which was associated with an impaired capability to differentiate into a mature neuronal lineage and reduced proliferation of NPCs.140 Furthermore, this defect in cellular proliferation was rescued by an inhibitor of glycogen synthase 3 (GSK3), which, as shown in animal studies, is an inhibitor of the Wnt pathway.111,142 Interestingly, the beneficial effects of lithium therapy in bipolar disorder treatment may be mediated by the inhibitory effect of lithium on GSK3.140,142
Studies on bipolar disorder have also shown that altered expression of brain-derived neurotrophic factor (BDNF) may play a role in the pathogenesis of this condition.143–147 The BDNF protein is a neurotrophic factor that acts on neurons of the central and peripheric nervous systems, which promotes survival of existing neurons and growth and differentiation of new neurons and synapses. Furthermore, BDNF has a role both in the early development of the central nervous system and in later regulation of neuronal plasticity in adulthood. 148,149 An iPSC study showed that the level of BDNF mRNA in iPSCs derived from 6 patients with bipolar disorder was lower than that derived from healthy donors, which may result in defects in regulatory mechanisms involved in neurodevelopmental and neural plasticity.150
Another study analyzed expression of microRNA-34a in iPSCs derived from patients with bipolar disorder.151 The iPSC-derived neuronal cultures showed an increase in microRNA-34a levels, compared with iPSCs from healthy donors. Higher microRNA-34a concentrations would then lead to an increase in the inhibition of the expression of ANK3 and CACNB3 genes, which have been previously categorized as genetic risk factors for the development of bipolar disorder, leading to changes in neuronal differentiation and synaptic plasticity.151 An increase in microRNA-34a levels was also recently observed in a study using NPCs from patients with bipolar disorder.152
Finally, evidence of defects in neurodevelopmental processes has also emerged in a study with iPSC cells from 3 patients with bipolar disorder, which showed alterations in a molecular cascade that controls spatial segregation of the central nervous system. In fact, neurons of patients with bipolar disorder increased expression of genes of ventral neurons, such as NKX2-1, while control-derived neurons increased levels of genes of dorsal neurons. These findings suggest that the presence of defects in the neural patterning and differentiation process in the developing nervous system may increase risk for onset of bipolar disorder.139 In addition, the genes related to transforming growth factor-β, which are implicated in early neuronal differentiation and proliferation, were expressed at lower levels in neurons of patients with bipolar disorder than controls.139
Overall, growing evidence indicates the presence of aberrations in several molecular mechanisms in neurodevelopment, rather than 1 mechanism, in iPSC models of bipolar disorder. Some studies reported links to defective Wnt signalling, which can lead to a decrease in the proliferation of NPCs and aberrant patterning in specific regions of the central nervous system, which can be partly rescued by lithium treatment.
Defects in neuronal morphology and interneuronal connectivity
Alterations in structural and functional brain connectivity have largely been reported and discussed in studies investigating the neurobiology of schizophrenia and bipolar disorder. 28,153 Brain imaging and postmortem studies have in fact shown morphological anomalies in brains of patients affected by schizophrenia, such as decreased connectivity between the thalamus and PFC and increased connectivity between the thalamus and sensorimotor cortex,154–157 alterations that can be detected during premorbid stages of the illness and that persist throughout the life of patients, according to longitudinal studies.158,159
Therefore, iPSC studies have examined alterations in the morphological features of neurons derived from patients with schizophrenia and bipolar disorder to characterize structural and connectivity defects.
Evidence from iPSC models in schizophrenia
One defective candidate mechanism is axon morphology and development, including axon guidance, which is defined as the process through which axons reach and identify their targets. 160–162 For instance, neurons derived from iPSCs of patients with schizophrenia showed a decrease in neurite length from a reduction in the expression of adhesion protein-coding genes, such as protocadherin (PCDH), neurexin (NRXN), and neural cell adhesion molecule (NCAM1) genes.163–165 These proteins are of primary importance in the regulation of interactions between neurons and in the trafficking of cell components.166
Other supporting evidence related to axon guidance defects involve the ephrin–Eph and Slit–Robo signalling cascades, which are known to influence the communication and targeting of axons from neurons of subcortical regions toward the appropriate cortical areas.109,110,112,113,160,167
For the ephrin–Eph pathway, studies in mice have shown that high concentrations of ephrin-A5 in the somatosensory cortex play a repellent role against innervation of limbic thalamic fibres.168 In fact, a decrease in ephrin-A5 causes excessive innervation of thalamic fibres to the sensorimotor cortex. 168 Studies of iPSCs found altered expression of ephrin-A ligands and receptors in cortical neurons and astrocytes derived from patients affected by schizophrenia.109,169
The Slit–Robo pathway is composed by Slit, a family of ligand proteins that act as a repulsive cue in axon guidance, and Robo, their transmembrane receptor; their several possible combinations during embryogenesis promote differentiation of the neural tube components.170 In mice models, mutations in this pathway are associated with alterations in thalamocortical and corticothalamic axonal targeting and corticocortical axonal pathfinding.171 In neurons derived from iPSCs of patients with schizophrenia, alterations in this pathway have been observed, such as a reduction in Robo2 and an increase in Robo3 and Slit2, which have been hypothesized to cause a decrease in cortical connectivity.110
Other alterations detected in iPSCs in schizophrenia that may lead to defective neuronal connectivity were those traced in axonal myelination, a process that is mediated by oligodendrocytes. 172 Two stem cells studies have shown that oligodendrocytes derived from patients with schizophrenia were altered in various aspects such as differentiation, morphological maturation, and viability,173,174 compared with iPSCs from healthy donors, which could lead to irregular myelination and defective strengthening of axonal pathways.
Different alterations have also been found in dendritic spine morphology and the branching process. For instance, a study involved an iPSC model with the 15q11.2 deletion, another genetic risk factor for schizophrenia.175 The 15q11.2 deletion leads to CYFIP1 haploinsufficiency, reducing the levels of cytoplasmic fragile X mental reduction (FMR) 1 interacting protein 1 (CYF1P1), which is implicated in synaptic functions on 2 levels.176,177 First, CYFIP1 interacts with the FMR protein, 178 which is a regulatory protein present in the neuronal cell body, proximal to dendrites and axons,179 that associates with polyribosomes to regulate the transcription of different proteins that operate at the synapses.180–182 The CYFIP1 protein also interacts with the Wiskott–Aldrich syndrome family member 1 and 2 (WAVE 1 and 2) proteins, involved in cytoskeletal remodelling. In an iPSC study, researchers focused on the effect of CYFIP1 deletion on NPCs from patients with schizophrenia carrying this mutation.183 These NPCs presented an altered expression of genes involved in cell cycle and cytoskeletal remodelling.183 Another study showed that neural rosettes generated from iPSCs with 15q11.2 haploinsufficiency displayed abnormalities in polarity and distribution of the adherens junction, caused by the reductions in CYFIP1 levels and, consequently, by altered interactions with WAVE 1/2.184 The results from these studies suggest an important role of aberrations in synaptic functioning and cytoskeletal remodelling in the pathophysiology of schizophrenia.
Defects in the formation of synapses have also been indicated as further morphological aberrations traced in components of the central nervous system in psychiatric disorders. Specifically, some iPSC studies have gathered evidence on how alterations in the expression of NRXN1, which encodes neurexin-1, and mutations in chromosome 15 may be linked to alterations in synapses morphology. Neurexin-1 is a presynaptic membrane protein that interacts with neuroligins (a type of postsynaptic membrane protein) to form synapses.185 Haploinsufficiency of NRXN1 has been categorized as a genetic risk factor for schizophrenia.186 In line with these results, an iPSC study found that haploinsufficiency of NRXN1 led to significant dysregulation of cell adhesion and neuronal differentiation, and reduced levels of synaptotagmins, neurexophilins, and vesicular trafficking proteins.187
Finally, dysregulation of synaptic remodelling has also been linked to alterations in brain connectivity and to the pathophysiology of mental health disorders. In humans, during the development of the PFC, synaptic density increases until childhood and reaches a peak before 10 years of age, after which it decreases during adolescence until the third decade of life.188 The decrease in synaptic connections is caused by a process called synaptic pruning, which is mediated by microglial cells.189 Alterations in synaptic pruning have been linked to schizophrenia. The most common age of illness onset for schizophrenia is in the early 20s to 30s,190,191 which coincides with the physiologic peak period of synaptic pruning. Postmortem studies have shown decreased density of postsynaptic elements in superficial PFC layers among patients with schizophrenia.192 These results are in line with an iPSC study that showed impaired synaptic pruning in vitro using neurons and monocytes derived from iPSCs of patients with schizophrenia.193
To summarize, the reported evidence in schizophrenia suggests a multifactorial and complex series of morphological defects in neuronal morphology, axon myelination and guidance, and synaptic modelling, which have all been implicated in the pathophysiology of this psychiatric disorder.
Evidence from iPSC models in bipolar disorder
Previous studies have revealed a series of alterations in iPSC models of bipolar disorder. One study focused on the differences recorded in the characteristics of cellular migration in iPSCs from patients with bipolar disorder.194 The time-lapse analysis showed that, among patients with bipolar disorder, migrating NPCs had a higher mean speed and abnormal directionality, with an increase in random trajectories, than cells from healthy donors.194 Furthermore, transcriptome analysis showed that expression of migration-related genes, such as cell surface receptors, were altered in models of bipolar disorder. 194 Defects in cellular migration in neuronal cells have also been linked to defects in neurodevelopmental processes.
Another study, using iPSCs from patients with bipolar disorder, showed further morphological alterations such as a decrease in the size of neurospheres and an increase in the length of their neurites.152 However, a different study reported a decrease in the length of neurites and in the number of synapses in bipolar disorder–derived cells, compared with control iPSCs.164
Two studies investigated abnormalities in bipolar disorder using 3D multicellular culture systems. One showed a decrease in size of the subventricular zone in bipolar disorder organoids, compared with those derived from healthy donors.152 In the other study, iPSC-derived human cortical spheroids of patients with bipolar disorder showed smaller size of spheroids, reduced proportion of neurons, reduced neural network activity, and decreased neuronal excitability compared with the 3D models obtained from healthy controls.195
These findings suggest that neurons induced from patient-derived iPSCs exhibit alterations in synapse- and dendrite-related phenotypes that are consistent with the defects in neural connectivity highlighted in bipolar disorder.
Dysfunctions in neurotransmitter systems
Dysfunctional neurotransmission has been regarded to play a primary role in the development of psychiatric disorders. One of the most accredited hypotheses underlying the pathophysiology of schizophrenia stems from the aberrations traced in different features of glutamate and GABA neurotransmission. Specifically, according to the glutamate hypothesis, symptoms in schizophrenia may be linked to reduced functioning of N-methyl-D-aspartate (NMDA) glutamate receptors and excessive glutamate signalling.196 This theory is in line with observations of the effects of NMDA receptor antagonists, such as ketamine, which can induce psychosis in healthy people.197,198 Furthermore, postmortem studies showed lower levels of NMDA receptors in brains of patients with schizophrenia, compared with healthy controls. 196 Since NMDA receptors drive the activity of cortical inhibitory interneurons, a reduction in NMDA receptor function, as recorded in schizophrenia, reduces the rate of GABAergic interneurons, resulting in disinhibition of pyramidal neurons.199
In addition, one of the most accepted and explored hypothesis to explain the symptoms observed in schizophrenia is a state of hyperactive dopaminergic signal transduction within the nigrostriatal pathways.97 Alterations in dopamine transmission have also been linked to the glutamatergic–GABAergic imbalance observed in schizophrenia, since the removal of tonic inhibition of dopaminergic neurons from frontal inhibitory neurons would then lead to excessive striatal dopamine release (Figure 1).37
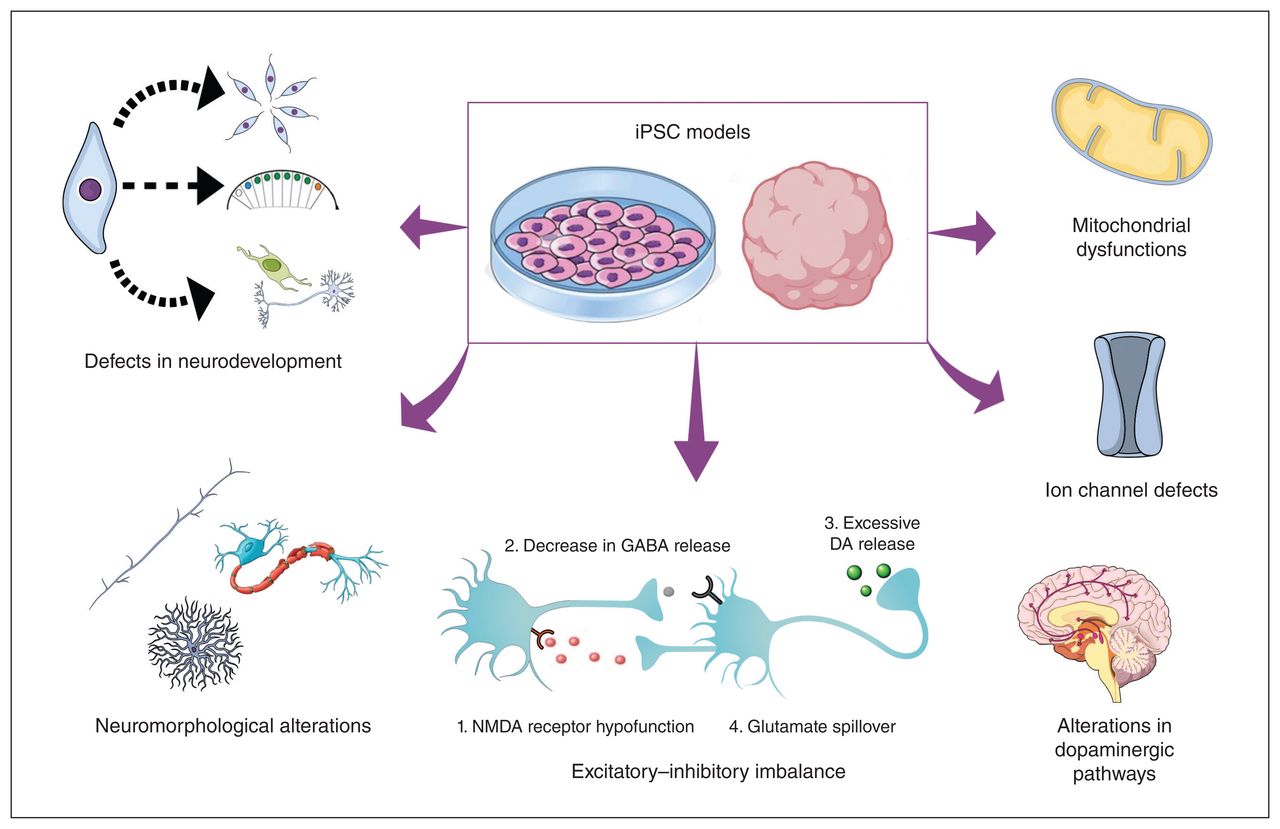
Pathophysiological alterations observed in iPSC models of schizophrenia and bipolar disorder. The development of 2- and 3-dimensional iPSC models has allowed the investigation of several, often interconnected, pathophysiological mechanisms that appear to contribute to the clinical presentation of schizophrenia and bipolar disorder. Research using iPSCs has reinforced the idea that these disorders share different neurobiological mechanisms, as shown by the detection of disturbances in neurodevelopmental processes (proliferation, segmentation, and differentiation), neuromorphological alterations (axon development, myelination, and dendrite branching), glutamatergic–GABAergic transmission imbalance (also linked to dysfunctions in dopaminergic pathways), and defects in ion channels and mitochondria, in both disorders. DA = dopamine, iPSC = induced plurpipotent stem cell, GABA = γ-aminobutyric acid, NMDA = N-methyl-D-aspartate.
Studies on the neurobiology of bipolar disorder have mainly focused on dysfunctions in monoaminergic neurotransmission in the central nervous system, also supported by the mechanisms of actions of pharmacological treatments and related to the clinical features of depression or mania.200 For instance, hyperactivity in the dopaminergic system has been linked to episodes of mania. In addition, alterations in glutamatergic and GABAergic levels have been recorded in bipolar disorder.200 Specifically, glutamate levels were found to be high in the postmortem brains of patients with bipolar disorder; the concentration of glutamate was decreased after valproate treatment, and GABA levels were increased after lithium treatment.201,202
Dysfunctions in neurotransmitter systems in iPSC models of schizophrenia
Several iPSC studies have confirmed aberrant glutamate signalling in schizophrenia. The main alterations observed were a reduction of the expression of different glutamate receptor subunits (i.e., GRIN2A, GRIN2B, GRIK1, GRIK2, GRM1, GRM7) and glutamate transporter genes in iPSC from patients with schizophrenia.92,110,165,167,203 Furthermore, a study showed that neurons derived from iPSCs of patients with schizophrenia presented alterations in the activity of the pituitary adenylate cyclase–activating polypeptide, a neuropeptide that acts on vasoactive intestinal peptide–expressing interneurons204,205 and enhances NMDA receptor activity.203 Furthermore, forebrain neurons generated from iPSCs derived from patients with both schizophrenia and DISC1 mutation had lower levels of DISC1 protein and deficits in synaptic vesicle release and synapsis morphology in glutamatergic neurons.206 In addition, it was shown that, by removing the DISC1 mutation through genetic engineering, the functionality of glutamatergic synapses was restored.206 Finally, iPSC studies have reported reduced density of excitatory postsynaptic elements in cultures of glutamatergic neurons derived from patients with schizophrenia, in accordance with genetic studies that have identified alterations in genes encoding postsynaptic proteins among patients affected by psychosis.207
Several studies have also shown defects in GABAergic transmission in iPSC models of schizophrenia. For instance, studies have shown alterations of GABA-synthesizing enzymes and differential expression of GABA receptor subunits in neurons derived from iPSCs of patients with schizophrenia. 92,109,110,203,208 In particular, an iPSC study conducted in a pair of monozygotic twins showed that brain organoids generated from iPSCs of the twin with schizophrenia had more GABAergic neurons than those of the healthy twin.109 Furthermore, genes related to the GABA receptor were over-expressed in neurons produced from iPSCs of the affected twin.109 Other studies on iPSCs have highlighted a downregulation of glutamate decarboxylase (GAD) genes in schizophrenia, which are involved in synthesis of GABA.92,203 This evidence could explain the decreased expression of GAD in the dorsolateral PFC of brains of patients affected by schizophrenia found in postmortem studies.209 Furthermore, a calcium imaging study using neurons derived from iPSCs of patients with schizophrenia showed that neurons from treatment-responsive patients (typical antipsychotic users) had a different response to GABA and glutamate than neurons from treatment-resistant patients (clozapine users); neurons from treatment-responsive patients had a decreased response to GABA and a normal response to glutamate, while those from treatment-resistant patients showed a normal response to GABA and an increased response to glutamate.210 Finally, other studies have found synaptic deficits associated with reduced expression of adhesion genes, such as PCDHs and neuroligin-2 (NLGN2), in inhibitory GABAergic neurons generated from iPSCs of patients with schizophrenia.163,208
Some studies have also shown defects in dopaminergic transmission in iPSC models of schizophrenia.161,211 For instance, a study using iPSC-derived dopaminergic neurons reported altered neuronal differentiation and maturation, with a reduced expression of tyrosine hydroxylase and βIII-tubulin, as well as a decrease in the density length of neurites and the absence of dopamine transporter expression.161 These findings are consistent with studies reporting defects in cortical neuronal growth and the functionality of the dopaminergic system, localized in the corticolimbic circuit, among patients with schizophrenia.97,212,213 Another study found that the maturation processes involving dopaminergic neurons were largely unchanged in cells derived from patients with schizophrenia, compared with healthy donors, but that the levels of dopamine production and release were higher among patients with schizophrenia,211 supporting the idea that an overactive dopaminergic system in areas such as the basal ganglia and the ventral mesencephalon may be at the core of this disorder.214
Overall, these results suggest that alterations in both glutamate and GABA neurotransmission may contribute to the pathophysiology of schizophrenia. In physiologic conditions, GABA interneurons suppress the excitatory activity from glutamate-expressing pyramidal neurons, resulting in an inhibitory feedback loop. In schizophrenia, inhibitory neurons are thought to receive attenuated input from excitatory neurons and thereby fail to effectively inhibit their target excitatory neurons, leading to neuronal hyperactivity in the corticolimbic system (Figure 1). These dysfunctions in excitatory–inhibitory cortical loops have also been linked to aberrations in electroencephalographic indices such as increased slow-frequency activity and defective synchronization of high-frequency activity.215–217 Further iPSC studies are required to improve our knowledge on the dysfunctions present in the dopaminergic system.
Dysfunctions in neurotransmitter systems in iPSC models of bipolar disorder
Few postmortem, imaging, and iPSC studies have highlighted defective glutamatergic transmission in bipolar disorder. 218–220 Specifically, 1 study, using iPSCs from patients with bipolar disorder, highlighted that disruption in circadian rhythm genes could be traced in NPCs and glutamatergic neurons. Furthermore, defects in expression of circadian rhythm genes were particularly evident in neurons of patients who were not responsive to lithium treatment, compared with iPSCs derived from healthy controls.220 Further iPSC studies focusing on glutamatergic transmission alterations in patients with bipolar disorder are required to improve knowledge on a possible imbalance in this neurotransmission system in this disease.
Regarding GABAergic transmission, an iPSC study compared microarray expression data in 4 siblings affected by bipolar disorder with 4 unaffected siblings at the neural progenitor stage, at 2 weeks (early stage) and at 4 weeks (late stage).221 Differences in gene expression between controls and affected patients changed during the 2 stages of differentiation. Specifically, there was an upregulation of GAD1 in neurons at the late stage among patients affected by bipolar disorder, compared with the neurons of healthy controls.221 Finally, in iPSC-derived NPCs and organoids of a patient with schizoaffective disorder, bipolar type (characterized by the presence of psychosis), an increase in GABAergic synapse-related genes and enhanced GABAergic specification was recorded, compared with the NPCs derived from a healthy twin.109
Dysfunctions in the dopaminergic system in iPSC models of bipolar disorder have been only rarely reported. The increase in dopaminergic activity in bipolar disorder has been hypothesized to be linked to the presentation of manic episodes and to the progressive loss of grey matter and synaptic density. For instance, a study showed that, in iPSC models derived from patients with bipolar disorder who did not respond to lithium, D1 and D2 dopamine receptor proteins were significantly increased, compared with models derived from healthy donors and lithium responders.222 Another study revealed that, in neural stem cell lines derived from patients with bipolar disorder, defects in the dopaminergic signalling pathway could be recorded during the early stages of neural development.223
Overall, the evidence from iPSC studies of bipolar disorder on the dysfunctions linked to the glutamatergic, GABAergic and dopaminergic systems or on the excitatory–inhibitory imbalance is still scarce and further studies are required to improve knowledge on the topic.
Defects in ion channel signalling
Genetic studies of schizophrenia and bipolar disorder have highlighted how the expression of different genes related to activity of voltage-gated cation channels can be linked to the development of these 2 psychiatric disorders.224 In fact, gene variants of potassium and calcium channels have been related to increased risk of developing schizophrenia, given the fundamental role of these channels in the regulation of several processes including neural plasticity, myelination, circadian neuronal rhythm, and energy regulation.56
Furthermore, an association between SNPs within calcium channel genes — such as CACNA1C, CACNA1D, and CACNG2 — and bipolar disorder has been consistently reported.57,225
Alterations in schizophrenia models
One study showed that iPSC-derived neurons from 13 patients with schizophrenia had different alterations in electrophysiological measures related to sodium channel function, such as an increase in membrane resistance and in the number of sodium current peaks.226 Alterations in the sodium channel function can severely affect the generation of action potentials and spike timing, which are critical processes for effective brain development and normal functioning. 226 The results were analyzed against a detailed clinical and cognitive characterization of the patients included in the study, showing that electrophysiological measures recorded in iPSCs could be employed to predict the severity of positive symptoms and cognitive deficits in these patients with schizophrenia.
Alterations in bipolar disorder and calcium signalling
Aberrant calcium signalling seems to be one of the primary features of the pathophysiological mechanisms of bipolar disorder. An iPSC study showed that neurons derived from iPSCs of patients with bipolar disorder presented alterations in the expression of calcium membrane receptors and calcium influx dynamics, compared with cells derived from healthy controls.152 Furthermore, 1 study analyzed RNA expression of NPCs produced from iPSCs of patients with bipolar disorder, showing both increases and decreases in expression of the different genes controlling the levels of calcium channels subunits.140 Finally, 1 study with iPSCs showed that, in neurons derived from patients with bipolar disorder, a hyperactivity in calcium signalling can be recorded, compared with cells from healthy donors.227 Furthermore, calcium activity was restored to physiologic levels in neurons treated with lithium, but only in cells derived from patients who were clinically responsive to lithium.227
According to the evidence from different studies, defects in calcium signalling seem to play a critical role in the pathophysiology of bipolar disorder, given the association of calcium dysfunctions with synaptic plasticity and homeostasis of the central nervous system. A better insight on the role of calcium in the pathophysiology of bipolar disorder would be beneficial for the development of new effective treatments.139
Mitochondrial dysfunctions
Alterations in mitochondria have been linked to aberrant synaptic maturation and remodelling because of their primary role in cellular respiration, regulation of intracellular calcium, and cellular apoptosis.228 In recent years, more studies have reported the presence of various defects in mitochondria in schizophrenia and bipolar disorder, such as alterations in calcium signalling and buffering, reduction in the production of adenosine triphosphate (ATP), and aberrations in mitochondrial size and density.229,230
Evidence in schizophrenia models
The 22q11.2 deletion has been associated with mitochondrial dysfunction.138,231–233 Li and colleagues232 produced neurons from iPSCs derived from patients with the 22q11.2 deletion who either had schizophrenia or did not present with any psychiatric diagnosis, showing that neurons derived from patients with this mutation had lower ATP levels and lower electronic transport chain activity than those derived from deletion carriers without schizophrenia, which expressed significantly higher electron transport chain activity than even neurons from healthy controls without the mutation. This suggests that, in the absence of the clinical manifestation of the disorder, a compensatory mechanism might be present in response to the alterations caused by the 22q11.2 deletion.232 Furthermore, one study that developed cerebral organoid from patients with schizophrenia showed deficits in mitochondrial functioning, basal oxygen consumption rate, and ATP production, compared with organoids derived from healthy donors.234
Evidence in bipolar disorder models
An iPSC study evaluated mitochondrial function in neurons generated from patients with bipolar disorder.227 This study showed that mitochondrial membrane potential in neurons from patients was increased, while the size of the mitochondria was reduced and overall neurons presented a generalized hyperexcitability, compared with iPSCs derived from healthy controls.227 Furthermore, researchers have theorized that the increased membrane potential of mitochondria might be consequential to the hyperexcitability status of neurons of patients affected by bipolar disorder.227 Interestingly, the neuronal excitability and mitochondria activity of iPSCs are influenced by treatments with lithium. As a matter of fact, alterations in excitability were rescued using lithium only in cell cultures derived from lithium-responsive patients. 227 In addition, lithium treatment improved the oxygen consumption rate and glycolytic rate only in mitochondria of NPCs derived from iPSCs of lithium-responsive patients with bipolar disorder, while valproate treatment improved these parameters in NPCs of lithium-nonresponsive patients with bipolar disorder.195 This evidence suggests that the efficacy of treatments could be assessed by recording changes in mitochondrial functioning in response to pharmacological treatments. However, this finding needs further replication since another study found that the mitochondrial membrane potential of NPCs was actually lower in iPSCs derived from patients with bipolar disorder, including both lithium-responders and lithium-nonresponders, compared with healthy controls.235 Furthermore, this alteration was not rescued by lithium or valproate, regardless of the clinical responsiveness to lithium of the patients.235 On the other side, one study that developed cerebral organoids from patients with schizophrenia and those with bipolar disorder did not detect any defects in basal oxygen consumption rate and ATP production of mitochondria in models of bipolar disorder.234
Understanding the evidence from iPSC studies
The current review provides a comprehensive overview on how iPSC studies hold promise for advancing our understanding of the main neuropathological alterations linked to schizophrenia and bipolar disorder.
For instance, iPSC models have been largely employed to investigate the effects of genetic variations, previously recognized as risk factors for schizophrenia and bipolar disorder, on cellular phenotypes.100,111,138,184,232 For example, allelic variations of the DISC1 gene in iPSC models have been associated with alterations within the Wnt pathway, leading to defects in neuronal proliferation, expression of fate markers,111 and formation of pre- and postsynaptic elements.206,236,237 In addition, variations in genes related to calcium channels — which can result in alterations in resting membrane potential of neurons — have been connected to both disorders, given the fundamental role of intracellular homeostasis of calcium in signalling processes, neural developmental pathways, and synaptic plasticity.25,238 Finally, genes involved in neuronal cell survival, proliferation, and differentiation have also shown a link to both disorders.109,239
Overall, strong evidence suggests common genetic factors for schizophrenia and bipolar disorder, but the genetic and phenotypic architecture of these disorders is highly complex, characterized by only a partial degree of overlap.25 In fact, the detection of shared genetic risk factors of the 2 disorders may be influenced by the subtyping of the patients considered, with type I bipolar disorder more strongly related to schizophrenia. 43 However, no iPSC study has been designed to explore the effects of shared genetic risk factors and mechanisms that may be at the core of the onset of one disorder rather than the other.
In addition to studies focusing on specific genetic factors, iPSC models have improved our understanding of the defects present in early neurodevelopmental processes in these 2 conditions.94 The iPSC studies reviewed seem to suggest that neurodevelopmental alterations in schizophrenia and bipolar disorder, such as the ones frequently found within the Wnt signalling cascade, cause a series of defects in proliferation, differentiation, and migration of NPCs (Figure 1).94,95,99,111
The presence of common neurodevelopmental aberrations may also reflect the correlates of trait-related features shared by both pathologies. For instance, a study investigating neuronal abnormalities using neuroimaging of adolescents who experienced an early onset of either schizophrenia or bipolar disorder with psychotic features showed that both groups presented cellular abnormalities, but these were more widespread in the schizophrenia group.240 Conversely, extracellular abnormalities could be traced only in adolescents with bipolar disorder.240 In line with this, Murray and colleagues38 hypothesized that patients with certain genetic predisposition to psychosis may be more likely to develop type I bipolar disorder by default, while the presence of additional altered neurodevelopmental genes or environmental factors would tilt the trajectory toward the onset of schizophrenia. Therefore, an interesting approach, only rarely employed in the iPSC studies reviewed, has been to focus on patients with shared clinical manifestations of the 2 disorders. Sawada and colleagues109 included iPSCs with 3 pairs of twins, 1 discordant for schizoaffective disorder (bipolar type) and the other 2 discordant for schizophrenia. Overall, deficiencies in the Wnt signalling pathway were traced in iPSCs of the patient with schizoaffective disorder, which led to a reduction in cell proliferation, a substantial shift toward the ventral fate, and an excitatory— inhibitory imbalance tilted toward GABAergic interneuron differentiation. 109 Furthermore, similar genetic alterations (mainly involving GABAergic transmission) were detected in both schizoaffective-(bipolar type) and schizophrenia-derived iPSCs.109 Therefore, this study provides an interesting view on the existence of a continuum between schizophrenia and bipolar disorder, tracing the possible shared mechanistic basis of psychotic symptoms through a dimensional rather than categorial approach. This is in accordance with 1 neuroimaging study that highlighted that patients with early-onset bipolar disorder (before age 18 yr) or bipolar disorder with psychosis showed neurodevelopmental alterations (as often recorded in schizophrenia) in cortical sulci morphology, compared with other patients with bipolar disorder or healthy controls.241 Other neuroimaging studies seem to report contrasting results since patients with schizophrenia and patients with bipolar disorder with psychotic features showed no shared alterations, supporting a certain grade of specificity in the development of schizophrenia.242,243
Schizophrenia and bipolar disorder present several trait-related features, but the neurodevelopmental alterations underlying the different state-related features of schizophrenia and bipolar disorder remain elusive. Furthermore, even within the same diagnostic category, differences in the iPSC alterations detected have been observed, probably owing to the heterogeneity of the patients with bipolar disorder selected for the studies.139,140 For instance, studies using iPSC from patients with bipolar disorder reported either an increase140 or a decrease139 in the levels of the PAX6 protein, affecting the normal fate of neuronal differentiation in bipolar disorder–derived iPSCs, or in the concentration of dopamine receptors, depending on patient’s responsiveness to lithium treatment.222
Overall, the results provided by the iPSC studies are heterogeneous but reinforce the theory that schizophrenia and bipolar disorder present several aberrations in neurodevelopment. However, the scarce number of studies using iPSC models of both disorders, using the same protocol, hinders the depiction of which specific mechanistic biological cascades determine the development of either schizophrenia or bipolar disorder at a later stage in life.
Patient-derived neurons display a series of morphological defects such as reductions in neuronal connectivity, neurite outgrowth, and synaptic markers (Figure 1). No specific phenotypic difference was detected in a study involving both schizophrenia-or bipolar disorder–derived neurons since both showed dendrite shortening and decreased synapse numbers.164 These results highlight the presence of common characteristics of the neurobiology of these disorders, which can be also linked to alterations in morphology and connectivity found in neuroimaging and postmortem studies.154,155
In addition, certain neuroimaging studies describe patients with bipolar disorder having functional connectivity similar to controls, while patients with schizophrenia being more similar to those with schizoaffective disorder, who might represent a phenotype intermediate between schizophrenia and bipolar disorder.239 However, tracing links between iPSC results, which could highlight the etiopathogenesis of these disorders, and results from neuroimaging studies, which may be influenced by the impact of the illness or the use of medication, remains challenging.
The various iPSC studies considered here reinforce the idea of a series of aberrations in neurotransmission. Specifically, the studies highlighted interspersed alterations in glutamatergic, GABAergic, and dopaminergic systems ranging from defects in receptors, in enzymes controlling synthesis of neurotransmitters, and in functionality of synaptic vesicles in schizophrenia.163,167 Furthermore, iPSC models have allowed detailed exploration of the excitatory–inhibitory imbalance, which is regarded as one of the core alterations present in schizophrenia and bipolar disorder.94,109 In schizophrenia, specifically, it seems that the excitatory–inhibitory imbalance may be a primary pathophysiological mechanism for the onset of the disorder, while the dopaminergic deficits emerge later in the cascade, when the illness is fully manifested.203 In this case, it is challenging to dissect how the shared defects in neurotransmission could be specifically linked to either schizophrenia or bipolar disorder. For instance, a study that compared iPSC of patients with either schizophrenia or bipolar disorder detected upregulation and downregulation of genes in glutamatergic and GABAergic neurons in both psychiatric models.164 However, alterations in gene expression, related to cell adhesion and neural function in these neurons, were more prominent in the schizophrenia model than the model of bipolar disorder.164 These observations suggest that neuronal dysfunction in both disorders can originate from either abnormal excitation or inhibition, depending on the patient’s background. Furthermore, development of schizophrenia may be related to the presence of a higher number of aberrations in genes involved in the formation of the neuronal system, compared with bipolar disorder models.25
Finally, 1 study investigated the involvement of mitochondrial dysfunctions in cerebral organoids from patients with schizophrenia and those with bipolar disorder.234 Specifically, the schizophrenia organoids showed deficits in basal oxygen consumption rate and ATP production compared with controls. 234 Conversely, no abnormal patterns were reported in the mitochondrial function of bipolar disorder organoids, potentially indicating a specific pathway of the neurobiology of schizophrenia. However, bipolar disorder showed specific dysregulation of gene expression in proteins controlling the interactions between the endoplasmic reticulum and the mitochondria. 234 This suggests that the altered mitochondrial dysfunctions recorded in both diseases may primarily result from distinct pathways, with schizophrenia arising more from dysfunctions in energy production, while bipolar disorder arising more from aberrant interactions between the endoplasmic reticulum and mitochondria.
Understanding the limitations of iPSCs and future prospects
Although the advent of iPSCs has allowed the creation of models that can efficiently track aberrations in molecular cascades present in neuropsychiatric disorders such as schizophrenia and bipolar disorder, some limits in literature and in the employment of this technique should be considered. In particular, iPSC models and results may vary depending on the culturing conditions, the type and number of cell lines employed, differentiation protocols, and freezing–thawing cycles of the cultures, hindering the reproducibility of results. 244 Furthermore, since neuronal development includes several steps in which multifactorial molecular pathways are involved, induced neurons cannot perfectly reproduce the complexity of processes that may be essential to the progression of the disease, such as neural migration, specification, or maturation, especially in 2D cell cultures.85
Finally, some limits regarding the artificial reprogramming of iPSCs (i.e., gene manipulations or exogenous treatments) should be taken into account. In general, the reprogramming process causes an alteration of the epigenetic landscape of the iPSCs from modification of the histones and DNA in specific sites.245 Studies that investigated the mechanisms underlying the reprogramming processes have also highlighted 2 major factors related to residual epigenetic memory of the somatic donor cells, specifically residual DNA methylation of their tissue of origin, namely that the genetic background of individual donors can influence iPSC features, which can be unrelated to any pathological state, and that the original cell type used for the extraction, which can affect or bias the differentiation capacity of the iPSC models, favours differentiation along their lineage of origin while restricting alternative lineages. 246 One approach to reduce the question of genetic diversity is to employ genetically related people under a family-based paradigm, including donors from members of the same family either affected by psychiatric disorders or not.140
To overcome these limitations, other cellular models using adult stem cells have been recently implemented for cell-based disease models. In 2010, multilineage-differentiating stress-enduring (MUSE) cells were isolated from the mononuclear cell fraction of the bone marrow; these are noncancerous, stress-resistant, and pluripotent endogenous stem cells.247 These cells can be easily isolated from human fibroblasts, bone marrow, and adipose tissue.247,248 Furthermore, the laboratory procedure to obtain MUSE cells, unlike that used for iPSC, does not involve gene manipulations or exogenous treatments, since MUSE cells are spontaneously capable of differentiating into phenotypes belonging to the 3 different embryonal sheets, enabling researchers to analyze patient-derived MUSE cells with their natural, original genomic background.249 To date, few studies have used MUSE cells, especially in the fields of neurology and oncology. 250–252 Interestingly, a study investigated the effects of mutations in the coding region of the IQSEC2 gene, which have been associated with intellectual disability and autism in MUSE cells derived from a patient with intellectual disability. 253,254 The patients’ cultures showed a reduced number of cycling cells and increased senescence compared with control cultures. In addition, deficits in neuroglial commitment and differentiation were detected in stem cells of patients.254
Another factor that may have influenced the heterogeneity of results discussed in the current review is that, from a clinical point of view, few study protocols provided a detailed clinical characterization of the sample.139,140,220,226,227 In fact, most studies did not specify the clinical profile of the enrolled participants, and provided only the general diagnosis, such as schizophrenia or bipolar disorder. Defining only the diagnosis poses a major limitation for 2 reasons, namely the overlap between schizophrenia and bipolar disorder in clinical presentation and the great heterogeneity within the spectrum of each psychiatric disorder. Therefore, common alterations traced in schizophrenia and bipolar disorder in neurobiological pathways may be linked to psychopathological or clinical dimensions rather than to a specific diagnosis. 27,32,33,255,256 Consequently, a detailed clinical characterization — including the content of psychotic symptoms, the temporal pattern of the association between psychotic symptoms and mood symptoms, and neurodevelopmental trajectories — may help to distinguish more clearly which patients showed substantial overlap in terms of genetic background, etiopathogenetic mechanisms, clinical presentations, and outcomes.
People presenting with the same diagnosis of either schizophrenia or bipolar disorder show a large heterogeneity in clinical presentation and extreme variability for the biological factors, neurobiological markers, and environmental variables associated with their pathology.257,258 This interindividual variability has hindered research in the field of schizophrenia and bipolar disorder since it has also affected the identification of the pathophysiological mechanisms underpinning their onset.
Therefore, a possible strategy to facilitate the advancement of knowledge in the field of the etiopathogenesis of these pathologies is trying to reduce interindividual heterogeneity by studying specific and more homogeneous subtypes of the aforementioned disorders. For instance, the diagnosis of deficit schizophrenia identifies a subgroup of patients with schizophrenia, presenting relatively homogeneous characteristics.259,260 In particular, people with deficit schizophrenia, compared with those with nondeficit schizophrenia, are characterized by poor premorbid adjustment, more frequently insidious onset, greater severity of negative symptoms and cognitive impairment, worse long-term outcomes, and higher frequency of neurologic soft signs.259 The latter constitute a broad range of subtle neurologic deficits, which have been strongly linked to impairments in neurodevelopmental trajectories.261
In the context of bipolar disorder, the distinction between rapid cycling bipolar disorder (i.e., ≥ 4 episodes of mood disturbance within 12 mo) and non-rapid cycling bipolar disorder is particularly promising to better characterize the clinical profile of patients. In fact, patients with rapid cycling bipolar disorder, compared with those with non-rapid cycling bipolar disorder, present different neurobiological alterations (e.g., a reduction in volumes of the ventrolateral PFC), an earlier onset, unfavourable outcomes in terms of number of hospital admissions, treatment response, and suicide attempts.46,262 Furthermore, patients with rapid cycling bipolar disorder have a higher rate of comorbidity with attention-deficit/hyperactivity disorder, which is also manifested at the level of shared polygenic risk, suggesting the presence of common alterations in neurodevelopmental processes.46,262 Therefore, it appears particularly relevant to use an approach based on the investigation of neurodevelopmental abnormalities in more homogeneous subtypes of the disorders, such as patients with deficit schizophrenia or patients with rapid cycling bipolar disorder.
Conclusion
Schizophrenia and bipolar disorder are psychiatric disorders with a complex genetic vulnerability for which animal models cannot recapitulate all disease-related pathophysiology. The advances in the development of iPSC models have allowed more detailed characterization of these 2 disorders, including a series of aberrations in neurogenesis, axon development, neurotransmission, and mitchondrial functionality. The complexity and heterogeneity of the results suggest that the development of schizophrenia and bipolar disorder may be linked to different and multiple types of alterations. Future research should consider using alternative stem cell models, such as MUSE cells, in addition to using a detailed clinical characterization to confirm a continuum between schizophrenia and bipolar disorder, and to achieve a more comprehensive understanding of the specific elements separating the neurobiology of these 2 disorders.
Acknowledgements
This authors acknowledge support from #NEXTGENERATIONEU and the Ministry of University and Research, National Recovery and Resilience Plan.
Footnotes
Competing interests: Silvana Galderisi reports consulting fees from Gedeon Richter; honoraria from Angelini, Boehringer Ingelheim, Gedeon Richter, Janssen, Lundbeck, Otsuka, Recordati, Rovi, and Sunovion; and participation on data safety monitoring boards with Angelini, Boehringer Ingelheim, Janssen, and Rovi. Giulia Maria Giordano reports consulting fees from Angelini. Armida Mucci reports consulting fees from Pierre Fabre, Rovi, and Boehringer Ingelheim; patents with Pierre Fabre; and participation on data safety monitoring boards with Angelini and Boehringer Ingelheim. No other competing interests were declared.
Contributors: All of the authors contributed to the conception and design of the work, drafted the manuscript, revised it critically for important intellectual content, gave final approval of the version to be published and agreed to be accountable for all aspects of the work.
- Received January 8, 2024.
- Revision received August 4, 2023.
- Revision received November 2, 2023.
- Revision received January 8, 2024.
- Accepted January 8, 2024.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is noncommercial (i.e., research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/
References
In this issue

{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.