

Abstract
In the field of schizophrenia research, as in other areas of psychiatry, there is a sense of frustration that greater advances have not been made over the years, calling into question existing research strategies. Arguably, many purported gains claimed by research have been “lost in translation,” resulting in limited impact on diagnosis and treatment in the clinical setting. There are exceptions; for example, we would argue that different lines of preclinical and clinical research have substantially altered how we look at antipsychotic dosing. While this story remains a work in progress, advances “found in translation” have played an important role. Detailing these changes, the present paper speaks to a body of evidence that has already shifted clinical practice and raises questions that may further alter the manner in which antipsychotics have been administered over the last 6 decades.
Introduction
The introduction of modern psychopharmacology through the 1950s revolutionized psychiatry, although it has also been argued that advances since then have been modest at best, with much of the research “lost in translation.”1,2
Although this may be the case, there are certainly numerous examples where advances in treatment have been “found in translation.” Arguably, antipsychotic dosing is one of these examples. Detailed here is a brief overview of how this story has unfolded; it remains unfinished but highlights several important messages. The first is familiar but often forgotten — little that we do in clinical medicine is written in stone; rather, it simply reflects the state of knowledge at the moment, with the inherent limitations of our tools and understanding. The second relates to research — rightfully, we place the greatest value on discoveries that fundamentally shift thinking and practice. Since the introduction of antipsychotics some 60 years ago, we’ve seen perhaps 2 of these in the pharmacotherapy of schizophrenia: the work that identified dopamine (DA), in particular dopamine D2 binding, as central to antipsychotic action, and the work on clozapine and its role in people with treatment-resistant schizophrenia (TRS). Notably, the discovery of antipsychotics was serendipitous,3–5 as was clozapine’s effect in people with TRS. In fact, clozapine was initially investigated as an antidepressant and was almost shelved after its release owing to unexpected deaths that were later linked to agranulocytosis.6 The point to be made, though, is that while we allot considerable weight and resources to the search for these major paradigm shifts, they are few and far between. In the meantime, there is the opportunity to make important and immediate advances in clinical care by ensuring that we are using what we have at hand as best as possible. What has happened with antipsychotic dosing seems to be one such example.
Antipsychotics and dosing: 1950–1990
As noted, the discovery of antipsychotics in the early 1950s was serendipitous, as chlorpromazine was initially being investigated for its potential presurgery anesthetic properties.3–5 Chlorpromazine itself was characterized by a heterogeneous receptor binding profile,7 and it was not until the late 1950s that attention turned to the relationship between antipsychotic efficacy and the monoamines. It was in the next decade that the focus turned specifically to DA, and thereafter DA D2 binding was identified as critical.8,9 In the meantime, the field witnessed the introduction of numerous other agents that would ultimately be termed low-potency conventional antipsychotics, characterized by heterogeneous receptor binding profiles (e.g., thioridazine, mesoridazine).
The findings related to DA D2 blockade became the catalyst for pharmaceutical companies to shift their focus to the development of highly selective D2 antagonists, premised on the notion that these would represent the “ideal” antipsychotic in terms of efficacy. Drugs like haloperidol and fluphenazine rapidly supplanted the older antipsychotics, although it turned out that the high-potency D2 blocking drugs were no more effective than their low-potency counterparts. Their much more selective D2 antagonism diminished the notable liability for cardiovascular side effects (e.g., postural hypotension) characterizing their low-potency counterparts, but at the same time led to a much higher risk of extrapyramidal side effects (EPS), which came to define the high-potency agents. One of the most notable results of the shift to high-potency antipsychotics was a dramatic increase in doses afforded by fewer cardiovascular complications. By 1980, daily doses were calculated to be 3.5 times the doses administered in early years,10 reflected in aggressive dosing strategies such as “rapid neuroleptization” (i.e., high antipsychotic doses over a short period to control acute psychosis).11
By the late 1980s and early 1990s, the field began to question the value of high antipsychotic doses based on available clinical evidence,12,13 although high doses continued to be used.14,15 What was missing was a biological framework around which this argument could be quantified.
Positron emission tomography, antipsychotics and D2 occupancy
Several key developments in the 1980s would fundamentally change the field. A seminal study involving clozapine established its clinical superiority in TRS,16 and efforts to account for its unique clinical and pharmacological profile led to the serotonin 2 receptor (5-HT2)/D2 hypothesis, which gained momentum through the 1990s and shaped the development of a new class of “atypical” or “second generation” antipsychotics.17,18 Positron emission tomography (PET) work examining antipsychotics and receptor occupancy in vivo was in its infancy at this point and initially focused on D2 occupancy, which was not surprising given the longstanding position that D2 binding was critical to antipsychotic efficacy.19 A variety of antipsychotics were examined at different doses, and the anecdotal finding that all patients with D2 occupancy in the range of 65%–85% responded well set the stage for “threshold occupancy” levels.20 Subsequent work highlighted the relationship between higher D2 occupancy and EPS, suggesting a threshold in the range of 74%–82%.21
A study from the same laboratory several years later confirmed that clozapine demonstrated low D2 binding in contrast to its notably higher 5-HT2 (and D1) binding, leading the authors to conclude “it can be speculated that D2 receptor occupancy per se is not the mechanism of action of clozapine.”22 Not necessarily incorrect, this finding fueled the notion that effective antipsychotics could be developed devoid of D2 binding.23 It also impacted drug development strategies; for example, interest in the role of 5-HT was so high at this point that efforts turned to the pursuit of selective 5-HT2 antagonists (e.g., ritanserin, M 100 907) as possible stand-alone antipsychotics, a line of investigation that would prove unsuccessful.24
Subsequent studies solidified the position that 2 thresholds existed in association with D2 occupancy, one associated with increased EPS (> 80%) and one linked to clinical response (60%–70%).21,25,26 Importantly, this work has also been carried out in patients with first-episode schizophrenia,27 which is clinically important because of evidence that antipsychotic response alters over the course of the illness.28,29
Translating D2 occupancy to antipsychotic dosing
To this point, antipsychotic dosing had been as much an art as a science, shaped by preclinical evidence and dose-finding studies seeking to establish a balance in terms of efficacy andtolerability and guided by information related to peripheral pharmacokinetics (e.g., terminal half-life [t1/2] and steady state levels).30 The use of plasma levels to identify thresholds for clinical response or side effects has not been particularly successful, in part because antipsychotics are metabolized through the hepatic cytochrome P450 enzymes, which are subject to numerous genetic and environmental influences, resulting in considerable intra- and inter-individual variability.31
The identification of thresholds based on D2 occupancy associated with optimal chance of clinical response, as well as increased risk of EPS, offered the most definitive means to date of establishing antipsychotic dosing guidelines; moreover, this could be immediately translated to clinical practice. Values being generated were not only useful in terms of individual compounds, but also provided the first empiric means of comparing doses between compounds, which was advantageous since antipsychotic switching is routine in clinical practice.32,33 However, this approach is not without its limitations. First, the thresholds represent guidelines and are not absolute; response can be observed at levels below 60%, just as nonresponse can occur at levels above 70%. In addition, not all antipsychotics can be classified according to this approach.27 Agents like clozapine and quetiapine, with their rapid dissociation values and short half-lives, reflect values that may not represent peak D2 occupancy levels at the time of scanning.22,34,35 Aripiprazole, with its partial agonist properties, also produces values that are difficult to reconcile with what is observed clinically (i.e., lower EPS profile despite high D2 occupancy).36 Notwithstanding such exceptions, it has been suggested that D2 occupancy levels may represent the most empiric means of establishing dose equivalents, at least in terms of antipsychotic activity given the central role D2 blockade appears to play in this symptom domain.37
Establishing dosing based on D2 occupancy has, in fact, been widely embraced in the literature, including preclinical studies38 and drug development studies.39,40 Moreover, there is evidence that this information has impacted clinical practice. Reports over the last decade have indicated that antipsychotic doses are actually declining,41,42 and treatment guidelines are now advocating much lower antipsychotic doses.43–45 Practice patterns have changed so dramatically that we now have to revisit what constitutes maintenance dosing, as we no longer routinely employ high doses in the earliest stages of treatment.46
D2 occupancy and dissociation constants
At the same time the imaging work involving dosing was being carried out, an alternative explanation to the 5-HT2/D2 hypothesis was evolving: the fast dissociation model.47 More specifically, studies involving cloned D2 receptors established that antipsychotics come off the D2 receptor at very different rates, with faster dissociation rates characterizing atypical antipsychotics.48,49 Values varied among the atypical antipsychotics as well, with drugs like clozapine and quetiapine among the fastest.50,51 This finding dovetailed with clinical evidence that the risk of EPS was the lowest with these compounds and was not dose-dependent.52
Even now the debate regarding which of these theories is correct continues when, in fact, they both have strengths and weaknesses. Neither, though, can lay claim to explaining clozapine’s unique clinical profile (i.e., superiority in refractory schizophrenia);53 evidence continues to confirm its superiority in this regard despite the development of agents that fit both models. At the same time, there is evidence that both models translate to diminished risk of EPS.52
What was critical about the fast dissociation model was that it countered the notion that we finally had evidence that D2 antagonism is not required for antipsychotic efficacy. In addition, at a molecular level it called into question the need for continuous D2 binding to establish and maintain antipsychotic response.
Central versus peripheral kinetics
Neuroimaging also provided the opportunity to elucidate central versus peripheral kinetics and, in the case of antipsychotics, examine this question from the standpoint of D2 occupancy. Historically, and even now, antipsychotic dosing has been based on the principles of peripheral kinetics, specifically t1/2, with the treatment goal of achieving and maintaining steady state levels.30 Single-dose studies examining both olanzapine and risperidone and involving healthy controls have shown that mean elimination half-lives for these 2 antipsychotics were 24.2 and 10.3 hours, respectively; in contrast, the average time to achieve 50% of peak striatal D2 receptor occupancy was 75.2 and 66.6 hours, respectively.54 Given the integral role of D2 occupancy in antipsychotic response,37 these findings called into question the longstanding strategy of establishing antipsychotic dosing patterns based on peripheral kinetics.
Transient D2 occupancy: A viable clinical option?
From the outset, antipsychotics have been administered in the form of regular and continuous treatment to minimize the risk of relapse. In fact, by the late 1950s depot antipsychotic formulations were introduced as a means of addressing nonadherence and ensuring gaps in treatment were avoided.55 It is still recommended that drugs with shorter half-lives be administered several times daily to ensure steady state levels, and, more recently, extended release oral formulations of several antipsychotics (e.g., quetiapine, paliperidone) have been developed and marketed, in part, based on their longer half-lives and avoidance of plasma level troughs.56,57
Only once in the last 50 years has this strategy been systematically evaluated. Driven by the substantial side effects associated with antipsychotics, “intermittent” or “targeted” antipsychotic therapy was introduced as a means of decreasing overall antipsychotic exposure.58–60 This approach entailed treating individuals when acutely psychotic and discontinuing treatment once they were stabilized. At the earliest signs of recurrence, antipsychotic treatment would be immediately reinitiated and again discontinued when the patient was stabilized. This strategy was ultimately abandoned based on clear evidence that such an approach resulted in increased relapse rates.61–63
“Extended” antipsychotic dosing
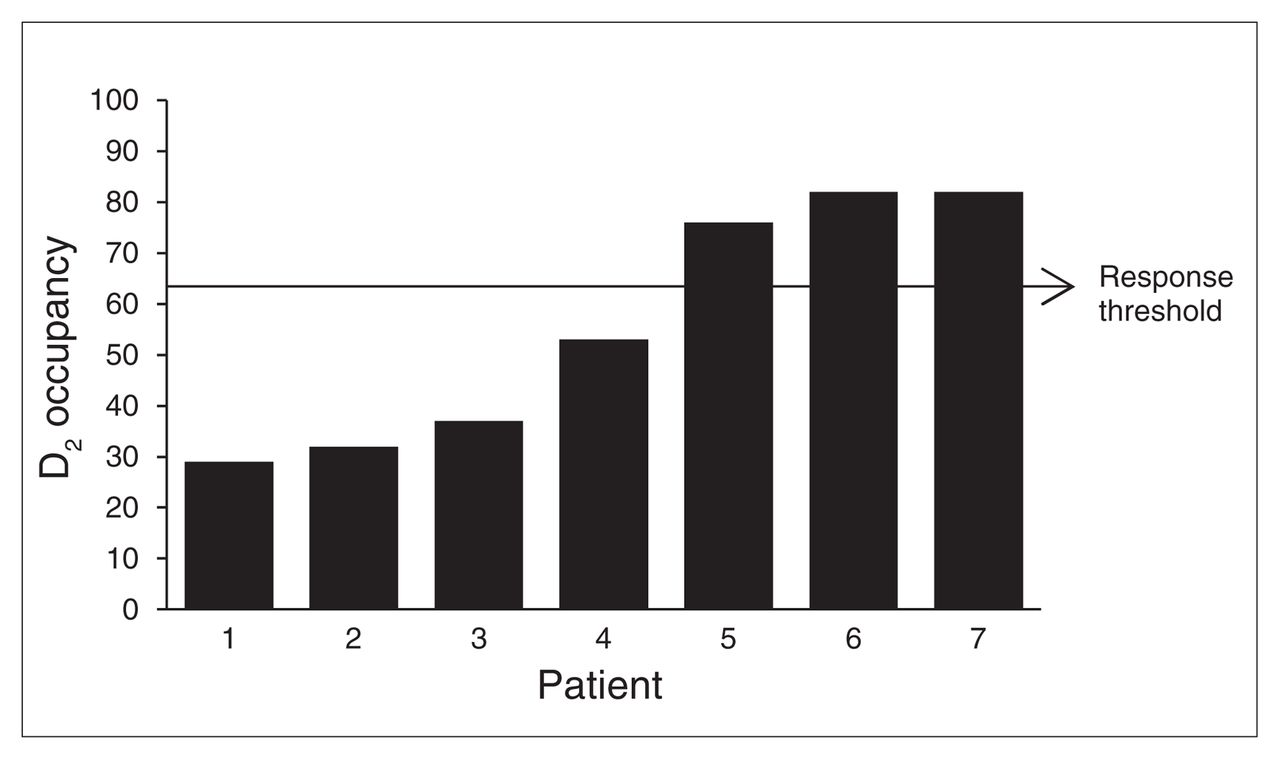
Three lines of investigation encouraged a re-examination of the axiom that continuous antipsychotic exposure is required to achieve and maintain response. At a molecular level, there was the preclinical work that led to the fast dissociation hypothesis.48,49 From the standpoint of imaging, PET data indicated that some of the newer antipsychotic drugs, clozapine included, were quite effective in the face of low D2 occupancy (which was interpreted as transient rather than uniformly low).26,34,35 Finally, work involving depot antipsychotic treatment offered at least indirect support for an approach that would allow for transient D2 binding, with earlier work indicating that fluphenazine decanoate, routinely administered every 2 weeks, could be administered in intervals as long as 6 weeks apart without compromising clinical response.64 Pursuing this line of approach with another antipsychotic, long-acting injectable risperidone, and doubling its recommended injection interval from 2 to 4 weeks, PET data confirmed that clinical response could be maintained in the face of D2 occupancy levels that fell below the 65% threshold by the time of the next injection (Fig. 1).65

Preinjection dopamine D2 occupancy levels in 7 patients taking long-acting injectable risperidone, with the injection interval doubled from the recommended 2 to 4 weeks. Only 3 patients had D2 levels above the threshold recommended to optimize clinical response, although none showed clinical deterioration. All had been on monthly injections for at least 4 months.
What could be taken from these findings? They did not negate the position that D2 blockade is required for antipsychotic response or that exceeding a threshold in the range of 65% was required to optimize clinical response. Evidence already confirmed that these features are necessary but not sufficient, reflected clinically in that subpopulation of individuals with psychosis who fail to respond to antipsychotic treatment, clozapine included.53,66 What the evidence also added was that high, sustained D2 occupancy is not required to maintain antipsychotic response.
This finding led us to suggest “extended” antipsychotic dosing, which argued for intermittent but regular drug administration (given the evidence arising from the earlier intermittent antipsychotic trials). This, of course, begged the question of what constitutes “regular.” The antipsychotic adherence literature was of little assistance at this point, as it routinely treats adherence/nonadherence as dichotomous and fails to address patterns of nonadherence.67 The PET data with drugs like clozapine and quetiapine indicated that the threshold did not have to be sustained over 24 hours, whereas the depot literature indicated that levels below this threshold were likely present for a matter of at least several days without compromising response. An open, proof-of-principle study was undertaken that would conservatively use a starting point of alternate day dosing. Participants stabilized on their antipsychotics were asked to take their antipsychotic medication every other day for 3 months, with a second 3-month window in which those who remained stable could reduce their dose to every 3 days. The study included 13 participants, 6 of whom completed the 6 months; only 1 participant, who stopped antipsychotic treatment entirely, relapsed.68 Among the remaining 6 participants, 5 withdrew consent over the course of the 6 months, although in no instance was withdrawl tied to symptom exacerbation; the sixth individual was withdrawn because of active substance abuse, an exclusion criterion.
These pilot data set the stage for a 6-month, double-blind, placebo-controlled trial69 in stabilized outpatients with schizophrenia comparing extended dosing (n = 18), here defined as alternate day dosing, with treatment as usual (TAU; n = 17). As in the first study, eligible individuals did not have to be asymptomatic to participate; it was only required that they be stabilized on their antipsychotics. Individuals in the extended dosing group did not show an increased risk of symptom exacerbation (Fig. 2) or hospital admission; more individuals in the TAU group required hospital admission over the 6-month interval.69

Total Brief Psychiatric Rating Scale (BPRS) scores for extended dosing (n = 14) and treatment as usual (TAU; n = 12) in participants completing the 6-month trial.
Is there a downside to continuous antipsychotic dosing?
If extended dosing is to be pursued, there is an onus to firmly establish its safety; larger trials are required to confirm the aforementioned findings. In addition, it is critical that we establish benefits over current practice. Intuitively, it seems straightforward that decreasing antipsychotic exposure by at least 50% will translate to gains for patients (over and above the fact that drug costs will be reduced by that same factor). In reality, however, taking medications on a nondaily schedule may be a challenge to adherence, which is an issue of concern for patients with chronic illnesses like schizophrenia.70 The aforementioned pilot study68 and double-blind study69 did not confirm clinical benefits, although they were both small and focused on efficacy. Going forward, investigations will need to pay closer attention to what subjective and objective benefits, if any, might be derived from this strategy across various domains (e.g., cognition, motivation, affect, motor).
This same question, though, can be approached from the opposite perspective: Is there a downside to continuous antipsychotic dosing?
Tolerance
Despite the numerous side effects of antipsychotics, current clinical practice stresses the importance of regular and continuous antipsychotic exposure and, unlike with medications such as benzodiazepines, dismisses the notion of tolerance.
In a recent commentary on this issue,71 we suggested that it may be necessary to revisit this position; our concerns arise from numerous and somewhat disparate pieces of evidence. In practice, clinicians talk of patients who, although initially responsive to an antipsychotic, lose the response over time,72–74 and the question of antipsychotic tolerance has been raised numerous times since their introduction.75–77 Simply put, the argument is one of tolerance in the face of continuous antipsychotic treatment, a phenomenon that has been described with other conditions that entail ongoing treatment (e.g., asthma).76
By the 1960s, preclinical models were able to identify physiologic changes supporting such a position, and in keeping with thinking at the time focus turned to alterations in biogenic amines.78,79 In and around this same period, there was evidence specific to chronic antipsychotic exposure indicating attenuation of drug-induced behaviours (e.g., stereotypy) in association with biochemical changes (e.g., number and affinity of D2 receptors).80 Much of this work was framed as hyper- or supersensitivity in the face of drug discontinuation81,82 and, as an aside, this became a model for tardive dyskinesia (TD).83
As early as 1980, a review posited that intermittent versus continuous drug exposure could possibly circumvent the effects of prolonged drug exposure (i.e., sensitization, tolerance).84 That differences exist as a function of intermittent versus continuous treatment has since been corroborated,85 including evidence of increased tolerance with continuous antipsychotic exposure.86 Most recently, 2 studies by Samaha and colleagues87,88 have provided further evidence of behavioural tolerance using measures sensitive to antipsychotic effects (amphetamine-induced locomotion and conditioned avoidance responding). In one study efficacy was attenuated with ongoing treatment, although response could be temporarily reversed with dose increases. Further, loss of response was associated with tolerance to haloperidol’s ability to increase basal DA and DA turnover, in combination with increased D2 receptor numbers, including proportion of D2 receptors in the high affinity state.87 Interestingly, on these same behavioural measures a second study demonstrated decreased efficacy with continuous versus transient haloperidol exposure, which was also reflected in biological measures that included striatal D2 receptor numbers and c-fos messenger RNA (mRNA) expression.88
Side effects
By the early 2000s, the atypical antipsychotics had largely replaced their conventional counterparts, with claims of clinical superiority and improved tolerability, including diminished risk of movement disorders, such as TD.89 Preclinical work in our laboratory pursued the TD question using a well-established rodent model of vacuous chewing movements (VCMs) as a proxy for TD in humans.90 Of note, the rapid metabolism of antipsychotics in rodents (e.g., olanzapine: t1/2 2–3 h in rodents v. 24 h in humans)91–93 provided an opportunity to model extended dosing, while administration of an antipsychotic in a depot formulation (e.g., haloperidol) or via osmotic minipump offered continuous exposure. Beginning with haloperidol, the prototypical high-potency conventional antipsychotic, it was demonstrated that risk of VCMs increased dramatically with continuous (depot; osmotic minipump) versus intermittent (daily subcutaneous injections) administration (Fig. 3).94,95 Turning to olanzapine, an atypical antipsychotic, it was established that continuous exposure via osmotic minipump resulted in high rates of VCMs compared with daily subcutaneous injections, which had VCM rates comparable to treatment with vehicle. Furthermore, the number of VCMs in the olanzapine minipump group was similar to that observed with haloperidol administered in a similar fashion.96 Taken together, these findings indicated that transient versus continuous antipsychotic exposure was associated with a markedly reduced risk of VCMs, even though the transient group demonstrated high D2 occupancy levels; the fact that exposure was not sustained seemed key to this diminished risk.

Vacuous chewing movements (VCMs) in rats exposed to continuous (pump) or intermittent (subcutaneous [SC]) administration of haloperidol (HAL) or vehicle (VEH).
Consolidating the evidence
Our understanding regarding antipsychotic dosing has been notably influenced by various lines of preclinical and clinical research, although the contributions have had both favourable and unfavourable consequences. For example, earlier work establishing the importance of D2 binding in antipsychotic activity proved to be a double-edged sword. On a positive note, this discovery culminated in the so-called high-potency conventional agents (e.g., haloperidol, pimozide), allowing us more clinically specific compounds that sidestepped problematic side effects linked to the older drugs with their nonspecific, heterogeneous receptor binding profiles. At the same time, the greater tolerability of high-potency agents, notwithstanding the increased risk of EPS (which could be treated with antiparkinsonian agents), resulted in a dramatic increase in doses, spawning an era of high-dose antipsychotic treatment captured in strategies such as rapid neuroleptization.
An argument against such high doses was gradually mounted based on clinical evidence; however, it was really neuroimaging that brought a level of sophistication that permitted the identification of specific dosing thresholds. Previously, the “neuroleptic threshold” hypothesis attempted to provide a clinical proxy based on side effects, specifically EPS,97 but it was neuroimaging that ultimately allowed for a cogent story linking D2 occupancy, clinical response and risk of EPS. Identified dosing guidelines based on this line of approach have translated to clinical practice, and evidence indicating that high doses98 and antipsychotic polypharmacy99 are still used is more likely a reflection of the sizable population with TRS.100
Although not an objective of the early PET work, findings related to the newer antipsychotics, in particular clozapine and quetiapine, in combination with preclinical receptor binding work, challenged the notion that high and sustained D2 occupancy is required to maintain antipsychotic response. This too had immediate implications for clinical practice, where antipsychotics are routinely administered in multiple daily dosing schedules shaped by their half-life. In fact, we now have various lines of evidence indicating that high and sustained antipsychotic dosing is not required, including data on single daily antipsychotic dosing with agents where multiple dosing is recommended based on peripheral kinetics; results from depot antipsychotic studies demonstrating that D2 occupancy levels fall below the recommended D2 occupancy threshold associated with clinical response; clinical findings that in the context of maintenance treatment depot injection intervals can be extended; and data showing that transient but finite gaps in oral antipsychotic administration (i.e., extended antipsychotic dosing) do not compromise clinical response. The implications of this work are, to say the least, controversial for a field in which antipsychotic nonadherence is an identified major contributor to poor clinical outcome101 and in the context of earlier evidence demonstrating that intermittent antipsychotic dosing increased the risk of relapse. It would not be unreasonable to ignore this evidence and default to the “safer” continuous antipsychotic dosing strategy used for decades were it not for the side effects of antipsychotics, which simply cannot be overlooked.
That said, more research, both preclinical and clinical, is needed to shed further light on antipsychotic dosing. In the last several decades, there has been closer attention paid to the question of how much, but it appears that we must also revisit the question of how often.71 There remain a number of important but, as of yet, unanswered questions. Is extended dosing an effective strategy, at least for some patients? Can clinical benefits be established with such an approach (e.g., decreased metabolic risk, diminished impact on negative and cognitive symptoms)? Conversely, is continuous antipsychotic exposure associated with adverse effects that can be mitigated or even avoided by regular but intermittent exposure (e.g., TD, morphological changes)? Does tolerance occur with continuous antipsychotic exposure? A recently published study on dose reduction and discontinuation in the context of first episode maintenance treatment touches on these very questions.102
At the very least, the evidence encourages us to keep an open mind regarding antipsychotic dosing. Our current “one size fits all” strategy is, in fact, at odds with our reconceptualization of schizophrenia as a heterogeneous group of disorders,103 which should encourage us to move beyond such a monolithic approach. There may well be individuals who do require antipsychotic doses continuously and, perhaps, even at high doses. Alternatively, there may be others for whom this is not the case and in whom response can be effectively maintained with an extended dosing type of approach while gaining the benefits of decreased antipsychotic exposure. Indeed, there is evidence that there may be a subgroup that does not require ongoing antipsychotic treatment.76
Are there strategies that may assist us moving in this direction? Randomized controlled trials will continue to be the gold standard, but we cannot ignore revisiting less sophisticated approaches with a new perspective that has us asking different questions. Naturalistic, observational studies that also include individuals who decline treatment, particularly from the onset of illness, represent a source of important information. Not surprisingly, in services already overburdened it is easier to monitor only those who buy into recommended treatment. Along similar lines, the more recent work examining trajectories of response/outcome may help us to distinguish subgroups.104–108
Conclusion
The challenges we face at present are allowing ourselves to accommodate such options and establishing means of accurately subtyping these different populations. However, if we do buy into the notion of individualized medicine, these possibilities seem somewhat less foreign.
Entertaining these sorts of ideas will require a paradigm shift. From the standpoint of drug development, for example, we are witnessing an upsurge in “long acting” compounds, both in the form of depot formulations and extended release oral agents. In fairness, the work involving depot antipsychotics is largely driven by nonadherence, a legitimate and significant concern in patients with schizophrenia. At the same time, a substantial proportion of patients are adherent and warrant the same attention regarding strategies that optimize response while minimizing side effects.
That we are even talking about antipsychotic dosing some 6 decades after the introduction of these agents is a telling reminder that we should not overlook what is in front of us, including practice patterns. There may be much more to be learned that can translate to enhanced clinical care, and we suggest that the search for new and more effective drugs be balanced with ongoing research to better understand and use those agents we presently have available.
Acknowledgements
This article reflects a talk given by Dr. Remington at the 2012 Canadian College of Neuropsychopharmacology (CCNP) Annual Meeting in Vancouver, British Columbia, in receipt of the 2012 Innovations in Neuropsychopharmacology Award. He expresses his deep appreciation to the CCNP as well as its membership for the award, and also wishes to extend special thanks to Drs. Shitij Kapur and Philip Seeman, who he has collaborated with over the years and whose ideas substantially contribute to this body of work. Each of the present authors is involved in work that is building upon lines of investigation discussed here.
Dr. Remington’s contributions in this area have been supported by the National Alliance for Research on Schizophrenia and Depression (NARSAD), Canadian Institutes of Health Research (CIHR), and Research Hospital Fund–Canada Foundation for Innovation, with additional support for specific projects through individual pharmaceutical companies. In addition, he receives research support through the University of Toronto’s Department of Psychiatry, the Centre for Addiction and Mental Health (CAMH), and the Campbell Family Mental Health Research Institute.
Footnotes
G. Remington is the recipient of the 2012 CCNP Innovations Award.
Competing Interests: In the last 3 years, G. Remington has received research support from the Canadian Diabetes Association, the Canadian Institutes of Health Research, Medicure, Neurocrine Biosciences, Novartis Canada, Research Hospital Fund–Canada Foundation for Innovation, and the Schizophrenia Society of Ontario and has served as a consultant or speaker for Novartis, Laboratorios Farmacéuticos Rovi, and Roche. G. Fervaha has been supported through an Ontario Graduate Fellowship. G. Foussias has been involved in research sponsored by Medicure and Neurocrine Bioscience and has served as a consultant for Roche. O. Agid has received research support from Pfizer and Janssen-Ortho and has served on advisory boards or speakers bureaus for Janssen-Ortho, Eli Lilly (Canada and U.S.), Novartis, Sepracor, and Sunovion. None declared by P. Turrone.
Contributors: G. Remington provided the framework for the review and wrote the article. All authors contributed to the development of the manuscript, reviewed it and provided final approval for its publication.
- Received September 3, 2013.
- Revision received October 16, 2013.
- Accepted October 23, 2013.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
Article tools



Related Articles
Cited By...
- No citing articles found.