

Abstract
Background: Tourette syndrome is a developmental neuropsychiatric disorder. Its etiology is complex and elusive, although an important role of genetic factors has been established. The aim of the present study was to identify the genomic basis of Tourette syndrome in a group of families with affected members in 2 or 3 generations.
Methods: Whole-genome sequencing was performed followed by co-segregation and bioinformatic analyses. Identified variants were used to select candidate genes, which were then subjected to gene ontology and pathway enrichment analysis.
Results: The study group included 17 families comprising 80 patients with Tourette syndrome and 44 healthy family members. Co-segregation analysis and subsequent prioritization of variants pinpointed 37 rare and possibly pathogenic variants shared among affected individuals within a single family. Three such variants, in the ALDH2, DLD and ALDH1B1 genes, could influence oxidoreductase activity in the brain. Two variants, in SLC17A8 and BSN genes, were involved in sensory processing of sound by inner hair cells of the cochlea. Enrichment analysis of genes whose rare variants were present in all patients from at least 2 families identified significant gene sets implicated in cell–cell adhesion, cell junction assembly and organization, processing of sound, synapse assembly, and synaptic signalling processes.
Limitations: We did not examine intergenic variants, but they still could influence clinical phenotype.
Conclusion: Our results provide a further argument for a role of adhesion molecules and synaptic transmission in neuropsychiatric diseases. Moreover, an involvement of processes related to oxidative stress response and sound-sensing in the pathology of Tourette syndrome seems likely.
Introduction
Tourette syndrome is a neurodevelopmental disorder characterized by motor and vocal/phonic tics persisting for longer than a year. The clinical phenotype of Tourette syndrome belongs to the spectrum of tic disorders.1 Moreover, most patients with Tourette syndrome present a variety of additional symptoms due to psychiatric comorbidities, including attention-deficit/hyperactivity disorder (ADHD), obsessive–compulsive disorder (OCD), autism spectrum disorder (ASD), affective disorders, anxiety disorders, impulse control disorders and personality disorders, implying an overlapping etiology.2,3 The tics usually emerge around the age of 4–6 years and are most severe after 5 years, albeit most cases improve during adolescence. The prevalence of Tourette syndrome in the general pediatric population ranges from 0.3% to 0.77%, and between 0.005% and 0.065% in adulthood.4 Other tic disorders are more common than Tourette syndrome and affect as much as 5% of the general population.3
Tourette syndrome and tic disorders in general have an important genetic component, with heritability estimated at 60%–80%.2,5 However, the clinical phenotype may be influenced by environmental, prenatal and perinatal factors; hormonal disturbances; and psychosocial stressors interacting with multiple genes.6–9 Based on the candidate gene approach and linkage studies, multiple genes have been suggested to be important in the etiology of Tourette syndrome.10,11 The protein products of these genes are involved in neurotransmitter signalling, synapse development, organization and functioning, differentiation of axons, cell adhesion and mitochondrial activity.11,12 However, the Human Phenotype Ontology database still lists only 2 genes, HDC and SLITRK1, as being involved in Tourette syndrome.13
Recent next-generation sequencing studies showed a complex genetic background of Tourette syndrome involving multiple interacting genes14 and a role of the rare variant burden in tic disorders.15 The available data suggest that de novo variants in approximately 400 genes contribute to Tourette syndrome risk in 12% of clinical cases.16 It is increasingly evident that rare pathogenic variants in a single gene cannot be responsible for a substantial fraction of Tourette syndrome cases.
Results of genome-wide association studies (GWAS) explain as much as 21% of Tourette syndrome heritability by polymorphisms, with a minor allele frequency (MAF) between 0.1% and 5%.8 The GWAS-based polygenic risk scores of tic disorders suggest that low-impact common variants, found also in the general population, contribute to the disease in a polygenic manner.17 Other GWAS have shown that Tourette syndrome is correlated with OCD, ADHD and major depressive disorder, diseases known to have an overlapping and highly polygenic background.18
Moreover, epigenetic mechanisms and gene regulation by noncoding RNAs have been proposed to mediate the influence of environmental factors on the genetic background of Tourette syndrome. Also, the role of rare noncoding variants remains largely unexplored in tic disorders. Thus, owing to the complex and heterogeneous genetic architecture of tic disorders, with common and rare variants in different types of genes associated with numerous biological pathways, the identification of susceptibility genes has been challenging.
We hypothesize that most cases of familial Tourette syndrome in the Polish population can be explained by an oligogenic inheritance of multiple variants. To verify this hypothesis we analyzed a group of families comprising people with Tourette syndrome as well as healthy family members using whole-genome sequencing to identify ultra-rare, rare and uncommon variants associated with Tourette syndrome.
Methods
Study sample
All patients with tic disorders were recruited from a single outpatient clinic and assessed by the same clinician specialized in tic disorders according to Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR, DSM-5) criteria. Patients were systematically interviewed using a semistructured interview based on the TIC (Tourette syndrome International database Consortium) data entry form.19 The prevalence of the most common comorbid disorders was evaluated and included ADHD; OCD; depression; anxiety disorders, including phobias, panic disorders, generalized anxiety disorder and separation anxiety disorder; oppositional defiant disorder; conduct disorder; and ASD. The list of obsessions and compulsions included in the Yale–Brown Obsessive Compulsive Scale (Y-BOCS) was used to establish the clinical spectrum of OCD. Each patient was carefully questioned about all the symptoms included in the DSM as the diagnostic criteria for the abovementioned comorbid disorders. Children and adolescents were assessed using the M.I.N.I. International Neuropsychiatric Interview for Children and Adolescents. Previous diagnoses of mental disorders that had been made in psychiatric clinics were accepted, and patients with severe psychiatric comorbidities were referred to a psychiatrist to confirm the diagnosis. All patients were from the Polish population, which is an ethnically homogeneous subgroup of White people.20 The study was approved by the Ethics Committee of the Medical University of Warsaw (KB/2/2007, KB/53/A/2010, KB/63/A/2018) and was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All participants or their legal representatives gave informed consent before inclusion in the study.
In case of a positive family history of a tic disorder, DNA was collected from all available affected relatives and healthy members of the proband’s family. Each patient was assigned to one of the following groups: Tourette syndrome, other tic disorders, or healthy controls.
Whole-genome sequencing
Participants’ DNA was extracted from peripheral blood leukocytes using a standard salting-out method21 or from saliva (Oragene DNA Self Collection Kit and Prep IT L2P Purification Kit, DNA Genotek Inc.). Whole-genome sequencing was performed by Novogene (Beijing, China) according to the following protocol. Sequencing libraries were generated using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs) following the manufacturer’s recommendations. DNA was randomly fragmented to 350 bp on average with a Bioruptor, and DNA fragments were size-selected with sample purification beads. The selected fragments were then end-polished, A-tailed and ligated with a full-length adaptor. The fragments were then filtered with the beads again. Finally, the libraries were analyzed for size distribution on an Agilent 2100 Bioanalyzer, quantified using real-time polymerase chain reaction and paired-end sequenced on an Illumina high-throughput HiSeq X Ten sequencer.
Fastq files were processed using the Intelliseq Germline Pipeline (https://gitlab.com/intelliseq/workflows) built with Cromwell (https://cromwell.readthedocs.io/en/stable/) according to GATK best practices. Variants were called with the GATK (4.0.3) HaplotypeCaller to yield genomic variant calling files (gvcf). All the code for file preprocessing and analysis is available at the GitHub repository of the project (https://github.com/ippas/imdik-zekanowski-gts) in the “Burden and family” section. Genotypes for the study group were obtained via the Hail run_combiner function (version 0.2.64, https://hail.is/).
Data analysis and filtering were performed in Hail (version 0.2.79). Multiallelic variants were split. The following variants were filtered out of the analysis: repeated and low-quality sequences (University of California, Santa Cruz RepeatMasker track ± 2 bp from each interval), loci with more than 90% gnomAD (v3) samples with a read depth (DP) of 1 or lower, variants with mean DP of 5 or lower, variants with mean genotype quality (GQ) of 50 or lower, variants that did not conform to the Hardy–Weinberg Equilibrium (HWE) (p < 0.05), variants with 3 or more samples with DP below 3, and variants with 30 or more samples with GQ lower than 30. The variants were annotated with gnomAD v3.1,22 combined annotation dependent depletion (CADD) scores,23 human phenotype ontology (HPO)13 and Ensembl Variant Effect Predictor (VEP).24 For each family, only variants that passed all quality filters for the whole group, had a CADD score above 10 and were within a gene locus based on Genecode v.3225 were taken for analysis.
Variant and enrichment analysis
The variants were analyzed according to their segregation pattern in each family (Appendix 1, available at www.jpn.ca/lookup/doi/10.1503/jpn.220206/tab-related-content) and were accepted if all patients within the family (with Tourette syndrome or tic disorder) had at least 1 alternative allele while none of the noncarriers had it. Only variants with the MAF in non-Finnish Europeans (NFE; based on gnomAD v 3.1) lower than the selected threshold (0.001, 0.01 or 0.05) were analyzed. Finally, the results were filtered to remove variants within genes that were called in only a single family.
A functional enrichment analysis was performed to investigate the biological relevance of the candidate genes using Metascape software.26 Lists of genes with variants co-segregating with the disease in at least 2–5 families were provided. A Log10(q) score lower than −2 was considered statistically significant.
For each observation, a probability of random occurrence was calculated (probability of observation; P(obs)). The chance of any variant(s) fulfilling the assumed criteria in any of the families was calculated using data from the NFE population in gnomAD (MAF or a probability of occurrence of ultra-rare/ rare/uncommon variant in a given gene), combined with probabilities of observing given segregation patterns in a given family or families. The results were adjusted for multiple testing using Bonferroni correction on the number of families, or number of combinations of 2–7 families (Appendix 2, available at www.jpn.ca/lookup/doi/10.1503/jpn.220206/tab-related-content).
Results
Participants
Our study sample included 17 multiplex families comprising 80 patients (40 with Tourette syndrome and 40 with other tic disorders) and 44 healthy family members (Table 1, Appendix 1 and Appendix 3, available at www.jpn.ca/lookup/doi/10.1503/jpn.220206/tab-related-content). Using whole-genome sequencing and variant analysis ultra-rare (MAF < 0.1%), rare (MAF < 1%) and uncommon (MAF < 5%) single nucleotide variants were identified.
Characteristics of included families
Cosegregation analysis
Analysis revealed 8282 uncommon variants present in all the patients from any single family (Appendix 4, available at www.jpn.ca/lookup/doi/10.1503/jpn.220206/tab-related-content), with 289 ultra-rare variants segregating according to the applied schemas and located in genes called in more than 1 family. Most of those variants were noncoding, including 4 variants located in genes encoding lincRNAs (LINC02306, LINC02763, LINC01414, LINC00298; Appendix 5, available at www.jpn.ca/lookup/doi/10.1503/jpn.220206/tab-related-content), with only 44 missense variants identified in protein-coding genes (Table 2).
Characteristics of ultra-rare, rare and uncommon variants*
Rare variant analysis
Analysis of rare variants segregating according to the applied schemas in at least 1 family revealed 97 variants with a CADD score above 20 (i.e., predicted to be within the 1% most pathogenic variants in the genome). There were 37 variants with a CADD score above 30 (i.e., predicted to be within the 0.1% most pathogenic variants in the genome (Table 3).
Rare and ultra-rare variants with the CADD score > 30 segregating with the disease in any single family
Three ultra-rare variants were identified in all patients from 2 families each (Table 4)
Ultra-rare and rare variants identified in 2 families each, sorted by MAF
Enrichment analysis
There were 121 genes with ultra-rare variants identified in at least 2 families; among them 13 genes with variants occurred in 3 or more families (Appendix 2). Of those 121 genes, 100 were amenable to gene enrichment analysis by Metascape, which showed 26 biological processes to be significantly enriched (Appendix 6, Supplementary table a, available at www.jpn.ca/lookup/doi/10.1503/jpn.220206/tab-related-content). As many as 60 genes were related to those 26 categories, with 38 genes involved in at least 2 significantly enriched processes.
There were 71 genes with rare variants identified in at least 3 families (Appendix 2), and Metascape could analyze 63 of those genes. In this set, 20 biological processes were found to be significantly enriched (Appendix 6, Supplementary table d).
Uncommon variants were identified in at least 4 families in 108 genes (Appendix 2), and Metascape could analyze 90 of these genes. In this set, 28 biological processes were found to be significantly enriched (Appendix 6, Supplementary table e).
The top significantly enriched processes, pathways, or cellular compartments from the analysis of all 3 data sets are presented in Table 5 and Appendix 7, available at www.jpn.ca/lookup/doi/10.1503/jpn.220206/tab-related-content).
Top significantly enriched categories (LogP value < −4) with variants associated with Tourette syndrome
Discussion
To obtain insight into the genetic architecture of Tourette syndrome we studied 17 large families from the Polish population at risk for Tourette syndrome by performing whole-genome sequencing followed by bioinformatic analyses. We posit that variants in genes present in all affected family members in at least 2 families are highly likely to have a role in the disease etiology. Most of the discovered variants were in noncoding regions, mostly intronic and some in 3′ and 5′ untranslated regions (UTRs). These variants may affect the gene in which they reside and may modify regulatory processes (e.g., by modulating noncoding RNAs) or their target regions.27 The CADD score, used here to prioritize genetic variants, gathers information about each variant from multiple sources, including conservation and epigenetic data.23 Therefore, a high CADD score indicates a likely high deleteriousness of a variant, including impactful mutations in noncoding, albeit possibly regulatory, regions. The enrichment analysis of the obtained gene lists revealed their association with several processes and structures, including cell adhesion and synaptic signalling. Some of them have been previously reported to be involved in Tourette syndrome etiology.
Variants cosegregating in 2 families
Three ultra-rare variants in genes not previously linked to Tourette syndrome (NLRP12, COL25A1 and IQGAP2) segregated with the disease in 2 families. None of the genes is pathogenic according to the American College of Medical Genetics and Genomics (ACMG) criteria. However, the identified genes could be considered valid candidate genes in Tourette syndrome owing to their involvement in neurologic processes. NLRP12 and COL25A1 have been associated with neurologic disorders and the IQGAP2-encoded protein could, through interaction with other proteins, influence calcium sensing and synaptic plasticity.
A missense variant in the NLRP12 gene was found in families G and I, one of the smallest in the study group (Table 1). The NLRP12 protein is involved in the inflammasome assembly and signal transduction; NLRP and its inflammasomes were upregulated in depressed and suicidal people.28 Polymorphisms in NLRP12 were associated with alcohol misuse and depression,29,30 and rare variants appeared to be causative of cold inflammatory syndrome31 and multiple sclerosis.32
The brain-specific membrane-bound collagen gene COL25A1 is expressed mainly in the hippocampus and the occipital lobe, with moderate expression in the frontal lobe and optic cranial nerve and low expression in the left cerebellum. The protein was first detected in amyloid preparations from Alzheimer disease brain and subsequently implicated in the pathogenesis of this disease.33,34 Polymorphisms in the COL25A1 gene were associated with antisocial personality disorder and substance dependence.35 Rare pathogenic variants in COL25A1 were reported in cases of congenital fibrosis of extraocular muscles.36
The IQGAP2 gene is a liver-specific member of the IQGAP scaffold proteins family. These proteins facilitate the formation of complexes regulating cytokinesis, cytoskeletal dynamics, intracellular signalling, cell proliferation and migration.37 IQGAP2 binds to calmodulin,38 and together with Munc13, forms a calcium sensor complex that controls short-term synaptic plasticity. Mutations in calcium/calmodulin-dependent serine protein kinase (CASK) were reported in autismspectrum disorders.39
Variants with a high CADD score cosegregating in families with Tourette syndrome
Among the variants with a high CADD score (> 30), 2 in aldehyde dehydrogenase genes and 1 in dihydrolipoamide dehydrogenase were present in 3 separate families. The aldehyde dehydrogenase (ALDH) superfamily plays an important role in pathways associated with development and detoxification; ALDH2 is crucial in the oxidation of aldehydes in the brain40 and is especially important in removing catecholaminergic metabolites (DOPAL and DOPEGAL) and the principal product of lipid peroxidation 4-HNE. Dihydrolipoamide dehydrogenase encoded by DLD catalyzes oxidative regeneration of lipoic acid cofactor. ALDH2 has been linked to neurodegenerative disorders (Alzheimer disease and Parkinson disease), but not with neurodevelopmental ones. However, a possible link between ADH/ALDH and drugs used in the management of akinetic and dyskinetic (resembling tics) movement disorders has been described.41 In addition, ALDH2 activation is associated with significant attenuation of depressive and anxiety-like behaviours in prenatally stressed rats, probably via modulation of various processes associated with inflammation, oxidative stress and apoptosis.42 In turn, pathogenic variants in DLD were reported in progressive neurologic deterioration.43 Proteomic analysis showed both ALDH2 and DLD as protein candidates that might be associated with susceptibility to stress-induced depression or anxiety and stress resilience.44 It could be hypothesized that rare variants in ALDH2 and DLD could hamper the aforementioned mechanisms and cause neurologic deterioration due to the sensitivity of the central nervous system to defects in oxidative metabolism.
Enriched processes
Most of the genes pinpointed in the cosegregation analysis in the families with Tourette syndrome were involved in neurologic processes, as indicated by the enrichment analysis showing that 12 of the 25 most significantly enriched processes (Figure 1) were specifically related to the nervous system (including neuromuscular processes). Many of these processes and compartments are interrelated (Appendix 8, available at www.jpn.ca/lookup/doi/10.1503/jpn.220206/tab-related-content). Moreover, many genes identified in our study that were associated with significantly enriched processes have previously been reported to be associated with other neuropsychiatric disorders (Table 6).
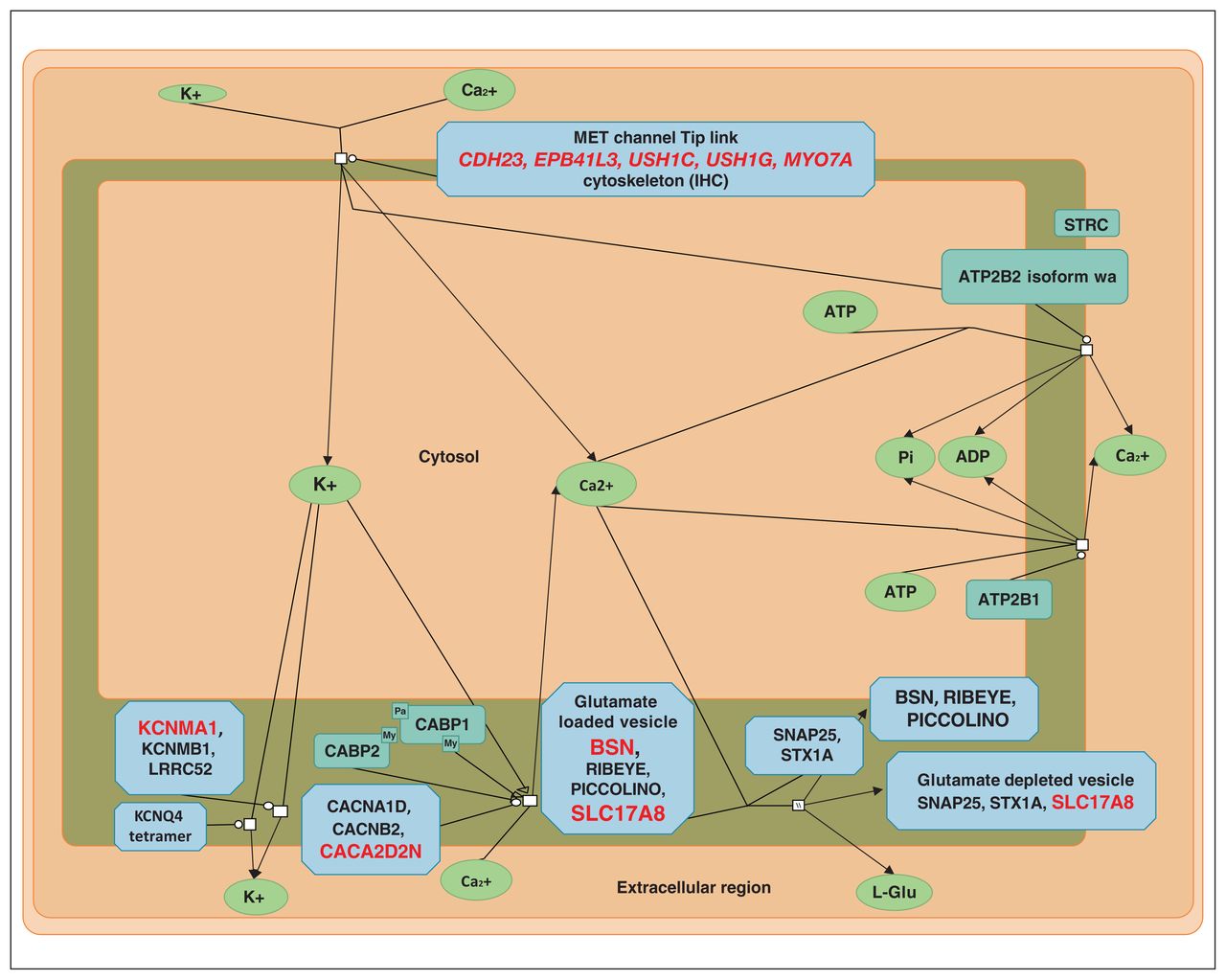
Sensory processing of sound by inner hair cells of the cochlea (modified from reactome.org). Proteins coded by genes with variants found in the present study are shown in red.
Genes identified as being associated with Tourette syndrome that were previously implicated in psychiatric and neurologic disorders
The top 2 enriched pathways with ultra-rare variants, cell junction assembly and cell–cell adhesion, although not neural-specific, relate closely with numerous genes already linked with Tourette syndrome (NLGN4, CDH23, CNTNAP2/CASPR2, and DPP6). Rare copy number variants (CNVs) in NRXN1 — a gene coding for the presynaptic cell adhesion molecule neurexin 1, which together with neuroligins,45 including neuroligin 4 (encoded by NLGN4), is involved in glutamatergic and GABAergic neurotransmission and synaptogenesis46 — have already been associated with Tourette syndrome.47,48 Moreover, NRXN1 deletions have been implicated in other neurodevelopmental and psychiatric disorders, including ASD and schizophrenia49,50
Variants in CNTN6 (overlapping CNTN4) and polymorphisms in TENM2 (rs147208935) and BTBD9 (rs9296249) were already associated with risk for Tourette syndrome.7,48,51 De novo variants in FN1 have been described in cases of Tourette syndrome.16,52 The genomic region encompassing PKP4 has been identified in GWAS analyses of Tourette syndrome and ADHD.14,53 ROBO2 has been identified as strongly associated with Tourette syndrome,14 and CDH23 was reported to be a candidate gene in a multiplex Tourette syndrome family.15 Our results are therefore in agreement with earlier analyses showing that cell adhesion and synaptic signalling is significantly related to Tourette syndrome etiology.14
A notable outcome of our enrichment analyses is that they identified a process that has, to our knowledge, not previously been mentioned in the context of Tourette syndrome: sensory processing of sound by inner (IHC) and outer hair cells (OHC) of the cochlea (Figure 1). The ultra-rare variants involved in sensory processing of sound by IHC of the cochlea were found in 5 families, and the uncommon variants were found in 8 (Table 7). Some families had a significant burden of variants in genes involved in this process. Two variants appeared to be pathogenic: SLC17A8(NM_139319.3):c.589–2A > C in the splice acceptor site and the missense BSN(NM_003458.4):c.3266G > C (p.Arg1089Pro) variant, both of which had a CADD score higher than 30. These variants are not found in any population databases (including gnomAD and 1000 genomes). They are located in conserved positions; have high CADD scores (within 0.1% of the most pathogenic variants), with evidence of pathogenicity according to ACMG criteria; and are predicted by many prediction programs to be pathogenic.
Variants in genes involved in the sensory processing of sound by inner hair cells of the cochlea*
SLC17A8 encodes vesicular glutamate cotransporter vGlut3, which accumulates glutamate in the synaptic vesicles of the sensory IHCs before releasing it into receptors of auditory nerve terminals.54 It is also present in striatal and serotonin neurons and is implicated in amphetamine-induced stereotypies in mice.55 BSN encodes bassoon presynaptic cytomatrix protein, which in IHCs is anchoring presynaptic ribbons essential in synchronous auditory signalling and, consequently, in normal hearing.56
The auditory system comprises descending efferent neural projections joining the medial and the lateral nuclei of the superior olivary complex (MOC and LOC, respectively) and the inner ear complex structure. The unmyelinated axons of the LOC project to the dendrites of auditory nerve fibres near the IHC afferent synapses. The role of the LOC system is still unknown; however, the presence of several neurotransmitters and modulators in its terminals (dopamine, acetylcholine, and GABA) suggests that it has complex functions. Moreover, it is known from animal models that, during a critical period of postnatal development, IHCs transiently receive cholinergic innervation, driving neurons in the auditory pathway to respond. This process is important for the normal maturation of synapses and circuits of the entire auditory pathway.57
The role of MOC is mainly to inhibit cochlear responses by decreasing the gain of the OHC amplifier. Several lines of evidence suggest that the MOC system plays an important role in the protection from trauma produced by overly loud sounds. The activity of the MOC toward the IHC input is also inhibitory during this developmental period and would control the excitability of the hair cells.
An important element of sound processing is also the prepulse inhibition (PPI), which is a measure of sensorimotor gating in response to a stimulus (pulse) being diminished when the stimulus is preceded by a smaller stimulus (prepulse).58,59 Sensorimotor gating is a normal protective mechanism in the brain that functions to gate or filter irrelevant sensory stimulation. Deficits in sensorimotor gating may result in stimulus overload and misinterpretation of sensory information. The hypothesis that the premonitory urges present in Tourette syndrome cannot be properly filtered and thus can induce abnormal, semivoluntary (so-called involuntary) movements like tics is in line with deficient PPI found in people with Tourette syndrome.58 Moreover, some tics can be triggered by diverse stimuli — auditory, visual, tactile or mental.60 A hypersensitization to various stimuli is a common clinical symptom in people with ASD. We also found this feature in 44.8% (74/165) of patients with Tourette syndrome, and almost one-third (22/74) had an abnormal response to sound.61
We identified 8 families in which variants of genes associated with the processing of sound by cells of the cochlea were found. There is some degree of phenotype–genotype interplay concerning overreactivity to different sensory stimuli. Six of 7 probands were oversensitized to touch (families D, G, H, I, J and T), and 1 proband additionally showed hypersensitization to sound (family H). There were no clinical data regarding oversensitivity to stimuli in 1 proband (family R) and remaining members of all analyzed families, including affected and healthy people.
Patients with ASD, OCD, or Tourette syndrome and misophonia, selective sound sensitivity syndrome or auditory hypersensitivity have been described previously.62–64 It has been proposed that these conditions may share pathophysiological development and etiological characteristics. Misophonia and Tourette syndrome could share abnormal activity of the limbic system, primary auditory cortex, and/or the autonomic nervous system.65,66 However, to our knowledge, no genetic basis for such a link has been proposed so far. We hypothesize that abnormal processing of acoustic stimuli, associated with the processes in the cochlear hair cells and superior olivary complex, and/or prepulse inhibition in our enrichment analyses may link the above-mentioned mechanisms and Tourette syndrome.
Limitations
Although we have analyzed variants co-segregating in 17 multiplex families with at least 3 patients each with tic disorders, the number of participants, including unaffected noncarriers, can be considered a limitation of the study.
Most of the genomic variants identified here are located in regions not coding for protein products; therefore, it is difficult to assess their impact on the processes in which the gene products participate. The putative causal link with the disease was based mainly on the cosegregation with the clinical phenotype, and additionally on the rarity of variants. The assessment of likely pathogenicity of the variants was based solely on in silico predictions. Additional functional studies, especially in the case of intronic or intragenic variants, will be needed, as different prediction tools could give substantially divergent rankings of variant severity.
We identified enriched processes and functions using the Gene Ontology (GO) database, although the biological processes are interdependent and particular genes (proteins or noncoding RNAs) may have roles in multiple processes. Moreover, the GO annotation is supported by various sources, and the majority of genes are assigned to terms based on computational predictions.
The aim of our research was neither functional nor translational, and further work to understand the genetic contribution to the clinical phenotype of Tourette syndrome and underlying dysfunctions at the molecular level is needed.
Conclusion
We identified putatively pathogenic genomic variants and molecular processes related to the etiology of Tourette syndrome in a group of Polish families. Three of these variants could influence oxidoreductase activity in the brain. Using enrichment analysis of the variant-bearing genes, we found evidence for a likely input of sensory processing of sound in the cochlea, in support of earlier reports of a hypersensitivity of a substantial fraction of patients with Tourette syndrome to diverse stimuli, including sonory ones. Other over-represented groups of genes with variants associated with Tourette syndrome were related to cell–cell adhesion, cell junction assembly and organization, synapse assembly and synaptic signalling, confirming earlier findings regarding the genetic basis of Tourette syndrome. Moreover, even if none of the identified variants is causal individually, our results support the concept of an oligogenic basis of Tourette syndrome and indicate that a burden of a large variety of rare and uncommon variants in genes implicated in various neurodevelopmental processes may be cocausally related to Tourette syndrome pathology. Further analyses using substantially larger groups of families, as well as individuals with sporadic Tourette syndrome and tic disorders, should be performed to confirm and expand our results.
Footnotes
↵* Share first authorship.
↵† Share senior authorship.
Competing interests: None declared.
Contributors: C. Zekanowski designed the study. P. Janik acquired the data, which J. Fichna, M. Borczyk, M. Piechota and M. Korostynski analyzed. J. Fichna and M. Borczyk wrote the article, which all authors reviewed. All authors approved the final version to be published, agreed to be accountable for all aspects of the work and can certify that no other individuals not listed as authors have made substantial contributions to the paper.
Funding: This work was funded by the National Science Center, Poland (NCN) project UMO-2016/23/B/NZ2/03030, and supported by PLGrid Infrastructure. J. Fichna is supported by the Polish National Agency for Academic Exchange, Bekker programme PPN/ BEK/2019/1/00452/U/00001.
- Received November 9, 2022.
- Revision received January 9, 2023.
- Accepted March 13, 2023.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is noncommercial (i.e., research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/
References
In this issue

{kind=link}
Article tools



Related Articles
Cited By...
- No citing articles found.